

哺乳动物的神经系统是由一系列不同的细胞组成的,这些细胞形成了复杂的功能联系。神经元亚型的细胞谱系规范、身份和功能背后的程序是由调节蛋白和rna管理的,它们以刻板的时间顺序协调步骤的继承。在中枢神经系统(CNS)中,运动神经元(MNs)通过整合皮层、脑干和脊髓(SC)向外周肌肉的信号传递,负责控制运动、呼吸和吞咽等基本功能。在引导祖细胞向MN方向发展的过程中,主要作用很大程度上归因于蛋白质因素。最近,一类在中枢神经系统中大量表达的调控rna——长链非编码rna (lncRNAs)的相关性已经压倒性地出现。lncrna驱动的基因表达控制是调控MN分化和功能的关键,其紊乱深刻影响神经元病理生理。在这里,我们发现了HOTaiRM1 (nHOTAIRM1)的神经元异构体的一个新功能,这是一个在SC中特异性表达的lncRNA。使用一个模型系统来概述脊髓MN (spMN)分化,我们发现nHOTAIRM1干预了MNs和中间神经元之间的二元细胞命运决定,作为一个促MN因子。此外,没有nHOTAIRM1的人类ipsc衍生的spMNs显示神经突生长改变,分支和连接数量显著减少。最后,当spmn中缺少nHOTAIRM1时,突触连通性和神经传递所必需的基因表达也会严重受损。在机制上,nHOTAIRM1与细胞质中的许多靶基因建立了直接和间接的相互作用,是MN生物学的一种新的转录后调节因子。总的来说,我们的研究结果表明lncRNA nHOTAIRM1对于MN身份的规范以及有丝分裂后MN的适当形态和突触活性的获得至关重要。
运动神经元(MNs)产生复杂的网络,将神经冲动从中枢神经系统(CNS)传播到外周组织,在那里它们被转化为肌肉收缩[1,2]。由于这些细胞在严重的退行性疾病中特别脆弱,因此最近对MN的发育和功能进行了特别关注[3]。在哺乳动物中,上神经元与下神经元建立谷氨酸能连接,将信息传递给目标肌肉。在下部神经网络中,脊髓神经网络(spMNs)代表了神经回路与骨骼肌的最终联系。
MN谱系的确定是由协调细胞类型特异性转录因子(TFs)组装的信号通路决定的,TFs触发MN特异性基因程序,最终抑制其他分化命运。TFs OLIG2和HB9是诱导spMN规范同时抑制中间神经元(IN)识别的典型分子[4,5]。长链非编码rna (long noncoding rna, lncRNAs)是一类在MN分化中也起核心作用的调节性非编码rna,它与信使rna (mrna)有几个共同的特征,但不被翻译成蛋白质[6]。它们作为基因表达的转录和转录后调节因子的功能,与它们同时与核酸和蛋白质相互作用的能力相结合,与大多数细胞事件有关[7]。这些独特的特征依赖于它们折叠成多个功能域的能力,实现一个模块化的组织,通过这个组织,它们结合和协调不同因素的活动。这使得lncrna具有非常多用途的分子,具有高度的作用特异性。
它们在中枢神经系统中普遍表达[8],在空间和时间上受到严格调控,这有力地证明了它们在神经发育过程和人类大脑进化中的意义[9,10]。此外,lncRNA失调对MN病理生理的深远影响是其参与协调细胞命运决定、细胞身份和该神经元亚型的功能[6]。尽管如此,很少有lncrna与MN的发育和活性相关[11],其主要功能是作为转录调控因子[6,7]。
我们之前将HOTAIRM1的神经元亚型(我们称之为nHOTAIRM1)描述为SH-SY5Y细胞在体外神经元分化过程中上调的lncRNA,被认为相当于神经元前体[12]。生理上,在脑组织中,nHOTAIRM1仅在脊髓(SC)中表达。根据这一观察,我们发现它在富集mn的腹侧SC谱系中积累,从人类诱导多能干细胞(iPSCs)分化出来[12]。这一证据促使我们探索nHOTAIRM1是否可能影响MN的生成和功能,最终了解其解除管制如何影响MN的生命周期。在这里,我们将基于基因组编辑的功能丧失方法应用于一个模型系统,该模型系统概括了spMN的分化,并确定了与MN发育和功能相关的关键nHOTAIRM1靶基因。我们发现nHOTAIRM1在MN生理学的多个方面都至关重要,从它们的形成(以INs为代价)到它们正确的形态和活性的实现。
根据基于谱系特异性诱导转录编程模块(NIL)的协议,iPSCs分化为功能性spmn[13,14]。通过对特定细胞标记物的qRT-PCR分析(图1B)来监测强力霉素(Dox)处理诱导的分化进程(图1A)。NANOG和OCT4被用作iPSC多能性标记物(D0),而iPSC向MN祖细胞(MNPs, D5)的转化是通过泛神经元标记物TUJ1和胆碱能MNs早期标记物MNX1(全文称为HB9)的表达来标志的[15]。在这个阶段,MNPs被解离并重新镀以进一步成熟。3天后(D8),细胞显示一致的ChAT表达,这是合成神经递质乙酰胆碱所必需的[16]。在第12天(D12),观察到LHX3和ISLET1的显著诱导,它们的协调表达引导细胞成为spmn[17]。在这一阶段,细胞群几乎完全由有丝分裂后的spmn组成,显示出功能性MNs的特性[14]。在这个分化集中,我们分析了nHOTAIRM1的表达(图1B)。在iPSCs中几乎检测不到,它在MNPs中逐渐被诱导,在spMNs中达到表达高峰(D12)。这种显著的表达增加表明nHOTAIRM1参与spMN编程,而其在有丝分裂后MNs中的高表达水平也表明其在spMN活性中的潜在作用。
图1:nHOTAIRM1前女友spMN分化及其在有丝分裂后spMN中的分布。
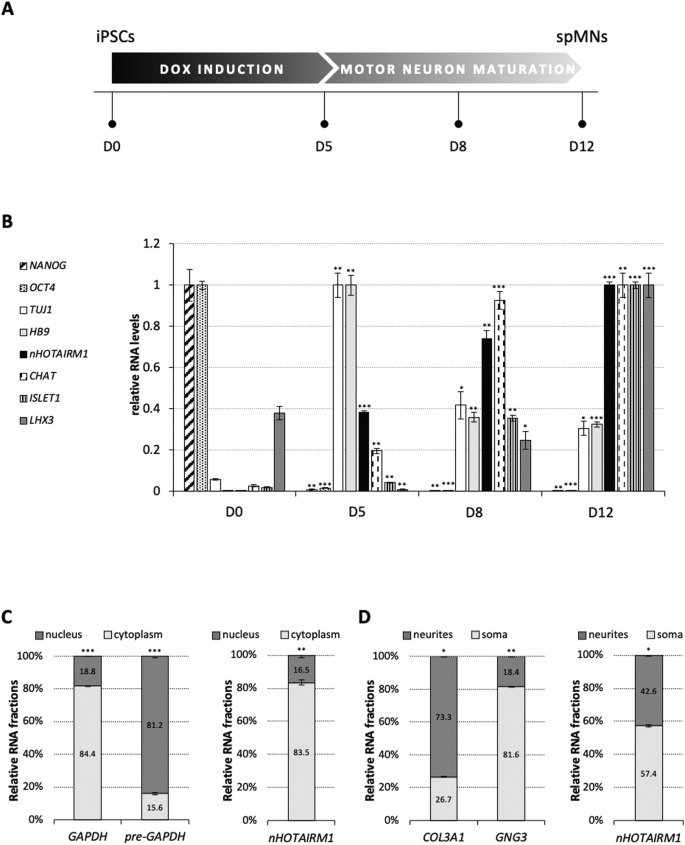
从iPSCs分化功能性spMNs的12天方案示意图。用多西环素(DOX induction, D0-D5)诱导细胞表达编程tf,然后再培养7天(D12)使spMN成熟。分化标记物和nHOTAIRM1在iPSC向spmn转化过程中的表达谱。qRT-PCR分析:多能性(NANOG, OCT4), MN祖细胞(TUJ1, HB9), MN规格(ISLET1),脊柱特性(LHX3),电生理活性(ChAT)。nHOTAIRM1表达式用黑色条表示。x轴表示分化天数。数据(平均值±SEM)以任意单位表示,相对于atp50mrna水平,作为内部控制。对于每个目标,将表达峰设为1。D0作为统计检验的参考样本。C spmn中nHOTAIRM1核/细胞质定位的qRT-PCR分析(D12)。左图:分别通过评估前GAPDH和GAPDH RNA水平来评估spmn的核/细胞质分离效率。总RNA归一化。数据(平均值±SEM)以GAPDH总量或GAPDH前水平的百分比表示。右图:nHOTAIRM1在spmn细胞核(深灰色)和细胞质(浅灰色)中的分布。总RNA归一化。数据(平均值±SEM)以占总nHOTAIRM1水平的百分比表示。nHOTAIRM1在spmn的体细胞和神经突中的分布(D12)。左图:分别通过评估GNG3和COL3A1 mrna的水平来评估体细胞/神经突从spmn分离的效率。总RNA归一化。数据(平均值±SEM)以GNG3和COL3A1总水平的百分比表示。右图:nHOTAIRM1分布于神经性(深灰色)和躯体性(浅灰色)隔室。总RNA归一化。数据(平均值±SEM)以占总nHOTAIRM1水平的百分比表示。各组统计结果如下:N=3个生物重复;* * * P≤0.05,P≤0.01,* * * P≤0.001,双尾学生的t检验。
为了揭示nHOTAIRM1在spmn中的功能,我们分析了它的亚细胞定位。以GAPDH和前GAPDH为对照(图1C,左图),spmn (D12)中的生化分离和表达分析显示,nHOTAIRM1主要存在于细胞质中(约83%,图1C,右图)。这表明其作为基因表达的转录后调节剂的潜在作用。接下来,我们加深了nHOTAIRM1在spMN细胞体(体细胞)和神经突中的分布。在体细胞/神经突分离后,分别通过检查GNG3和COL3A1定位进行评估[18,19](图1D左图),lncRNA的相对富集量被量化(图1D右图),显示其在两个区域的发生率相当,细胞体也由核对应物贡献。
这种分布是通过结合RNA荧光原位杂交(RNA FISH)和免疫荧光(IF)的成像分析证实的。图2显示,nHOTAIRM1(红色点表示)在体细胞中富集,但也在神经突以及远端轴突段(黄色箭头)中积累。这些发现暗示了nHOTAIRM1在多种功能中的潜在作用,如神经突生长、轴突运输、轴突驻留mrna的局部翻译或突触活性。
图2:可视化nHOTAIRM1有丝分裂后spmn的亚细胞定位。
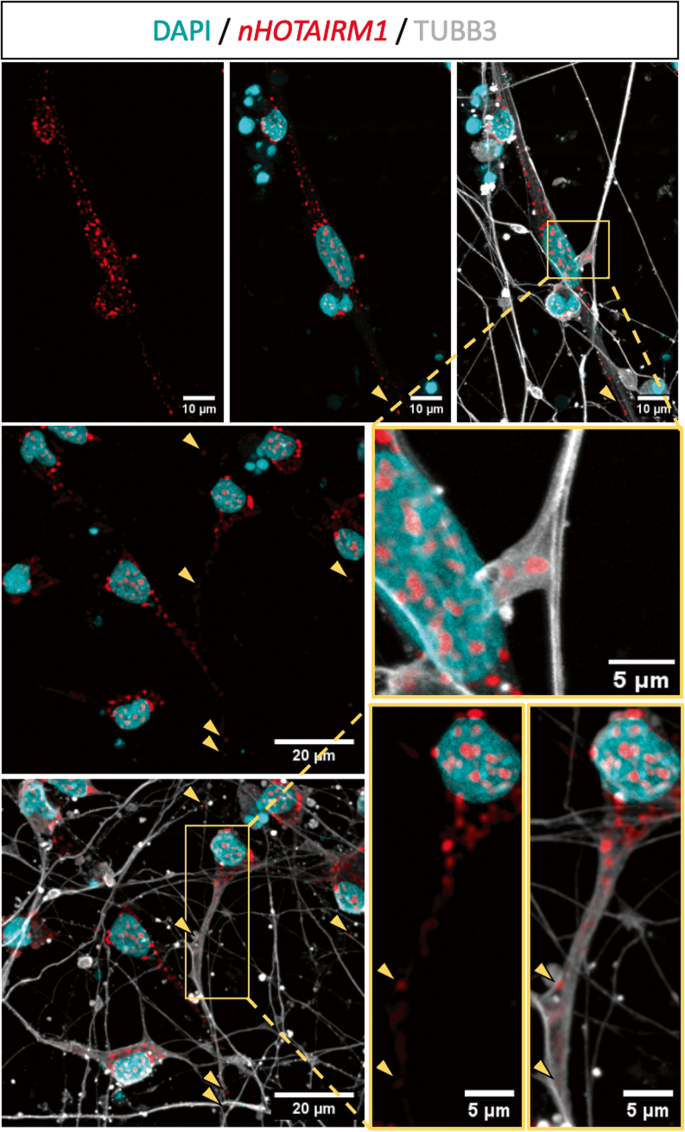
在ipsc衍生的spmn中,nHOTARIM1的RNA FISH(红色斑点)与TUBB3 IF(灰色斑点)结合(D12)。细胞核反染成青色。方形插入是数字放大的黄色边框面板。黄色箭头指向远端轴突节段的nHOTAIRM1点。比例尺单位显示在每个面板上。
为了解码nHOTAIRM1的功能,通过对野生型(WT)和HOTAIRM1敲除(KO) spmn (D12)进行转录组学分析,鉴定了其靶基因。KO iPSC系是通过CRISPR/Cas9基因组编辑获得的,通过一个过早的poly-A信号[20]阻断HOTAIRM1基因的转录,该信号插入其第一个外显子的5 '区,位于Zenbu报道的注释转录起始位点峰的下游(https://fantom.gsc.riken.jp/zenbu/)(图3A)。获得了两个独立的HOTAIRM1 KO纯合iPSC克隆(KO#1和KO#2)。图3B显示,在两个KO克隆的整个分化过程中,lncRNA的诱导量急剧减少,在分化的KO#1和KO#2 spmn中分别减少了约87%和96% (D12)。
图3:nHOTAIRM1来电显示KO部分。
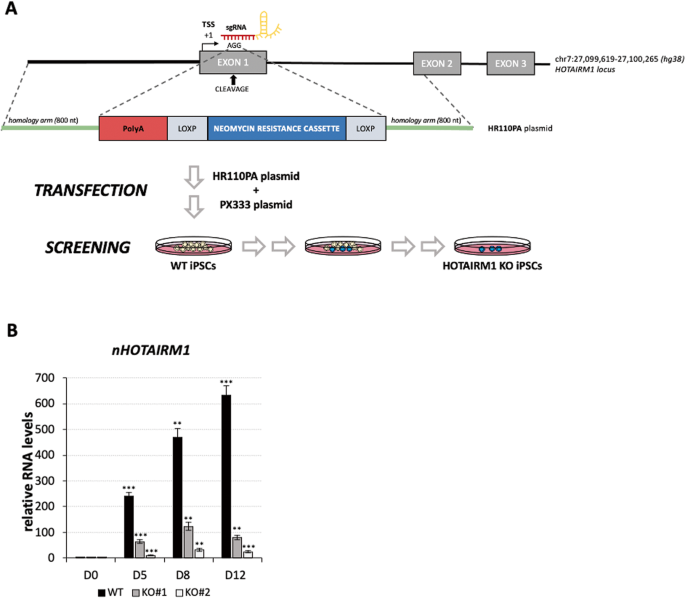
HOTAIRM1基因编辑实验示意图。设计了两个sgrna(由质粒PX333携带)靶向HOTAIRM1位点的外显子1。HR110PA质粒携带polyA信号、选择盒和同源臂,用于通过DNA重组进行同源定向修复。细胞操作示意图如下。野生型(WT)和nHOTAIRM1 KO (KO#1和KO#2) spmn (D12)中nHOTAIRM1表达的qRT-PCR分析。数据(平均值±SEM)以任意单位表示,相对于atp50mrna水平,作为内部控制。N=3个生物重复;*P≤0.05,**P≤0.01,***P≤0.001,双尾Student 's t检验,简称D0为参考样本进行统计检验。
在WT和KO#2 spmn的三个独立生物重复上进行的RNA-Seq分析的数据挖掘显示,两个样本都是均匀聚类的,并产生了许多基因的读计数(补充图1A, B)。我们在至少一个样本(数据集1)中检测到16504个基因表达,其中,|logFC | > 1且调整后p值< 0.05的基因被认为是显著差异表达。应用这些过滤器,我们在KO spmn中发现了1,887个上调基因和654个下调基因(数据集1),这些基因被认为是真正的nHOTAIRM1靶点(补充图1C-E)。使用WebGestalt R工具(http://www.webgestalt.org/)对差异表达基因列表进行基因本体过表示分析。从这个测试中,只有FDR < 0.05的基因集被认为是显著富集的。对于上调基因,“Biological Process”、“Cellular Component”和“Molecular Function”结构域的分析显示了高度异质的本体注释,与MN生物学没有严格的关系(补充图2-4,图A)。查询KEGG和REACTOME通路数据库也得出了类似的适应症(补充图5、6,图A)。当应用于下调基因时,所有这些计算检查表明,值得注意的是,“生物过程”本体显示了与MN (i)分化、(ii)形态和(iii)活动(数据集1和补充图2B)明确相关的注释,这些注释指导了随后的分析。
为了首先验证RNA-Seq分析,通过qRT-PCR分析了WT和KO spmn中10个上调和下调基因的亚群。选择调整后p值< 0.05且显著性值在logFC?2.72 ~ logFC 7.65之间的候选数据。值得注意的是,它们都遵循RNA-Seq分析中突出的表达趋势(补充图7A, B)。
随后的功能研究集中在属于七个下调的生物过程的三个主要基因组,涉及MN生物学的基本方面。它们包括过度代表的基因参与:(i) MN分化(“脊髓细胞分化”和“中枢神经系统神经元分化”);(ii)形态学(“神经元投射引导”、“轴突发生”和“轴突引导”)和(iii)活性(“突触组织”和“化学突触传递的调节”)。
为了探索nHOTAIRM1在神经元细胞命运决定中的作用,我们比较了WT和KO iPSCs在转化为spmn过程中与分化相关的靶基因的表达谱(图4)。我们在KO细胞中证实了与神经元发育相关的基因的表达模式的改变,如TF和辅助因子SALL1[参考文献[21]]和LMO4[参考文献[22]],它们在发育中的大脑皮层、核受体RORa、它识别中枢神经系统中的兴奋性神经元[23]和跨膜受体EPHB1,这对于调节依赖接触的细胞间相互作用很重要[24](图4A)。我们还验证了与SC或MN发育相关的基因的失调(图4B),如DLL4(在SC的神经元亚型规范中起作用)[25],PROX1(腹侧SC模式的调节剂)[5],LHX4(腹侧MN分化所需)[26]和DCC(与MN迁移相关)[27]。这些基因的表达谱在iPSCs形成KO spMNs的过程中发生了改变,在D12时减少了54%至90%。这表明nHOTAIRM1是运动神经元发育所必需的。
图4:例神经相关基因的表达分析沿着co的Nal微分和的反转nHOTAIRM1KO iPSCs进入有丝分裂后spMNs。
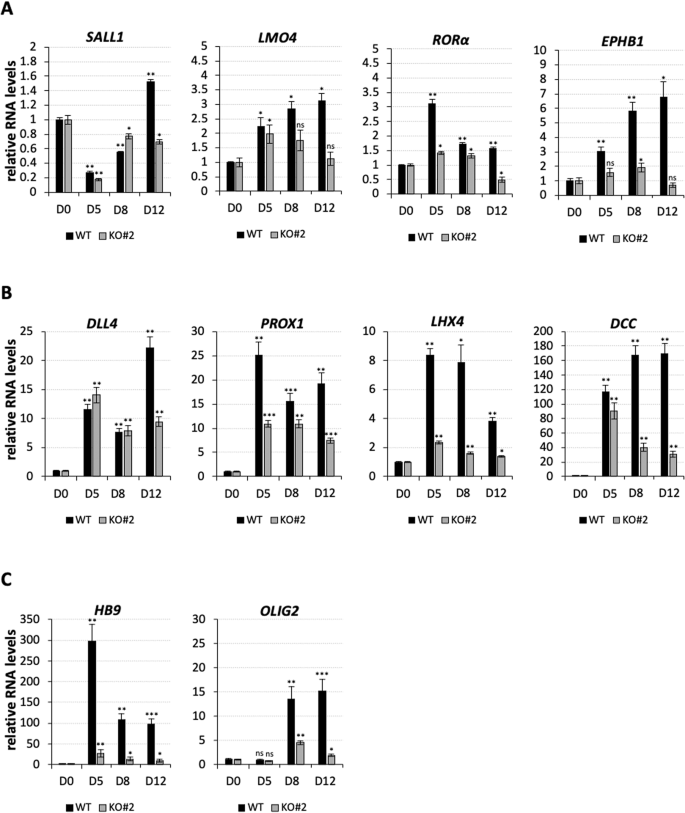
通过qRT-PCR分析,nHOTAIRM1 spMNs (KO#2克隆)沿WT或KO分化(从D0到D12, x轴)与神经元发育(A)和与SC或MN发育(B)相关的基因(每个面板上方显示)的表达谱(A, B)。nHOTAIRM1 spMNs (KO#2克隆)的HB9和OLIG2基因沿WT或KO分化方向(从D0到D12, x轴)进行qRT-PCR分析。数据(平均值±SEM)在所有面板中以任意单位表示,相对于atp50mrna水平,作为内部控制。对于每个目标和每个细胞系,D0时的表达量设为1。N=3个生物重复;*P≤0.05,**P≤0.01,***P≤0.001,双尾Student 's t检验,简称D0为参考样本进行统计检验。
随着KO spMNs从iPSCs分化,HB9和OLIG2也出现了一致的下调(图4C),这两个基因与获得替代神经元命运至关重要。HB9基因是发育中的SC中MN的选择性标记物[4,28],对于确定MN身份和抑制in特征至关重要。同样,它的调节因子OLIG2也会启动MNPs成为MNs[29],并抑制IN的同一性[5]。在此基础上,我们想知道nHOTAIRM1是否能够以牺牲INs为代价,积极地控制MNs的产生。为了回答这个问题,我们分析了胆碱能神经元标记物ChAT[30]和主要MN标记物ISLET1[参考文献[4]]在iPSCs转化为spmn过程中的表达。我们发现它们的表达水平在KO iPSCs的分化过程中急剧降低(图5A),突出了nHOTAIRM1在spMN生成中的相关性。IF分析证实了这一发现(图5B和补充图8),其中我们观察到nHOTAIRM1耗损后CHAT + /ISLET1 + MN细胞数量显著减少约42%。在这种情况下,缺乏nHOTAIRM1会损害spMN分化,我们询问是否进行了另一种细胞命运。鉴于nHOTAIRM1调节HB9的表达,这是MN和IN细胞命运之间的分子转换,我们想知道lncRNA的缺失是否可以部分模仿HB9的缺失。
图5:MN和hb9依赖性IN基因的分析nHOTAIRM1KO。
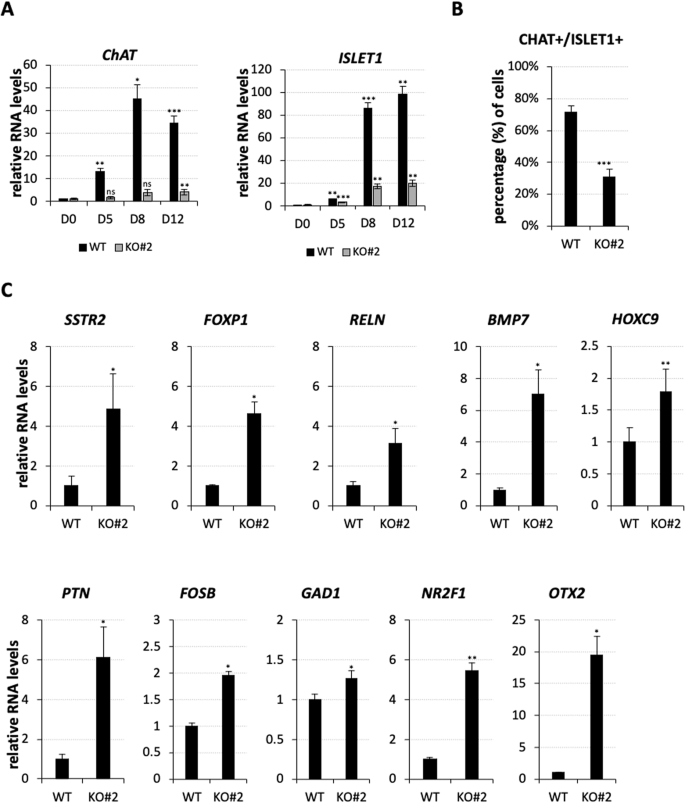
通过qRT-PCR分析,nHOTAIRM1 spMNs (KO#2克隆)的ChAT和ISLET1基因沿WT或KO分化(从D0到D12, x轴)的表达谱。数据(平均值±SEM)以任意单位表示,相对于atp50mrna水平,作为内部控制。D0时表达量设为1。N=3个生物重复;*P≤0.05,**P≤0.01,***P≤0.001,双尾Student 's t检验,简称D0为参考样本进行统计检验。B免疫染色定量检测WT和nHOTAIRM1 KO (KO#2) spmn中CHAT + /ISLET1+细胞,以细胞总数的百分比表示(D12)。N=3组,从9个不同的野区随机选取60个细胞,分别进行WT和KO分析。*** p≤0.001。C qRT-PCR验证,在WT和nHOTAIRM1 KO (KO#2克隆)spMNs (D12)中,HB9依赖性in基因的表达(每个面板上方显示)在HB9 KO MNs和nHOTAIRM1 KO spMNs中与WT spMNs相比显著上调。数据(平均值±SEM)以任意单位表示,相对于atp50mrna水平,作为内部控制。对于每个目标,在WT spmn中的表达设为1。N=3个生物重复;* * * P≤0.05,P≤0.01,* * * P≤0.001,双尾学生的t检验。
为了验证这一点,我们首先交叉了nHOTAIRM1 KO spMNs和HB9-null突变小鼠诱导的MNs中显著上调的基因列表[31]。我们通过qRT-PCR在KO spmn (D12)中验证了在INs中表达的10个共享基因的特征(图5C)。综上所述,这些结果表明,nHOTAIRM1通过控制HB9的表达,调节了INs和MNs之间的二元命运决定,并有利于后者。这一证据得到了特定IN标记基因的明确证实,即CALB2、SST、ETV1、LHX1、LAMP5、PVALB和VIP,我们发现这些基因在nHOTAIRM1缺失时上调(补充图9A)。
神经突的生长取决于促进功能性神经元连接形成的几个基因的活性[32]。有趣的是,比较转录组分析揭示了一系列在KO spMNs中下调的关键参与神经突生长的基因。我们重点研究了NrCAM、UNC5A、ROBO1、DCC、SEMA3E和SEMA6D这6个基因,发现它们富集于“神经元投射引导”、“轴突引导”和“轴突发生”3个本体基因类别。特别是,在体外与神经突生长活性密切相关的NrCAM[33]和参与轴突寻路[34]的UNC5A的表达分别下降了约53%和95%(图6A)。在KO spMNs中,通过与SLIT配体相互作用参与轴突引导的受体ROBO1的表达减少了约66%,而编码NETRIN-1受体的DCC的表达减少了约80%,具有促生长活性[37](图6A)。此外,SEMA3E和SEMA6D的表达在KO spMNs中分别减少了约66%和46%,SEMA3E和SEMA6D属于信号蛋白家族,在发育中的神经系统中指导轴突引导[38]。这些数据表明,nHOTAIRM1调节了几个与神经突网络组装有关的关键基因的活性。
图6:小波变换和nHOTAIRM1有丝分裂后spmn。
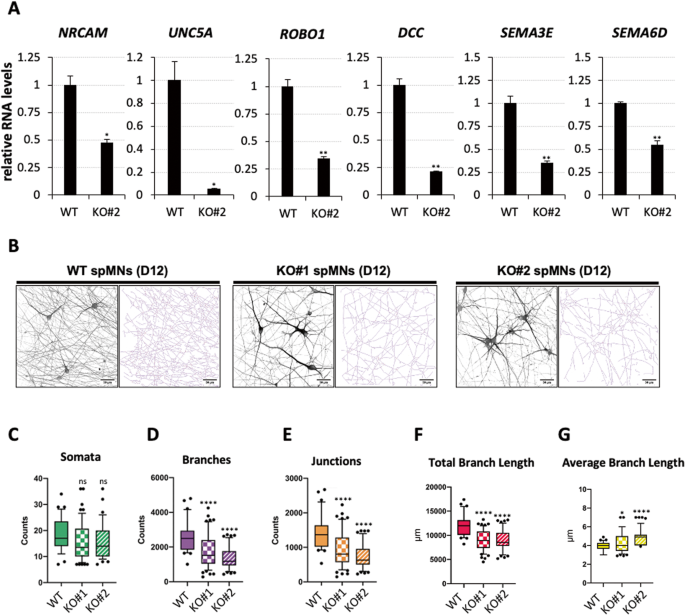
WT和nHOTAIRM1 KO (KO#2克隆)spMNs (D12)中与神经突生长和分支相关的基因的qRT-PCR分析(每个面板上方显示)。数据(平均值±SEM)以任意单位表示,相对于atp50mrna水平,作为内部控制。对于每个目标,在WT spmn中的表达设为1。N=3个生物重复;* * * P≤0.05,P≤0.01,* * * P≤0.001,双尾学生的t检验。B . WT和nHOTAIRM1 KO (KO#1和KO#2克隆)spmn中具有代表性的神经突网络采集和骨架化(D12)。比例尺单位显示在每个面板上。在WT和nHOTAIRM1 KO (KO#1和KO#2克隆)spmn (D12)中,每次获取的体细胞数(C)、分支数(D)、连接数(E)、总分支长度(F)和平均分支长度(G)。箱形图中的水平线表示中值;方框从每组值分布的第25到第75个百分位数延伸;黑点表示异常值。箱形图表示N=3个独立生物重复的值分布。总的来说,每个重复分析了10-20个212.3 μm × 212.3 μm的采集数据。*P≤0.05,****P≤0.0001对应单因素方差分析多重比较检验。
接下来,我们将KO#1和KO#2 spMNs的形态与WT spMNs的形态进行了比较,以验证有缺陷的分支表型是否反映了这些分子分析。由WT或KO spmn组成的神经突网络图像(图6B)按照文献[39]的描述进行分析(补充图10)。从所有样本中细胞数量相等开始(图6C),计算总分支数(图6D)和连接数(图6E),并测量总分支长度(图6F)和平均分支长度(图6G)。与WT相比,在两个KO神经突网络中发现分支和连接数的计数在统计学上显着减少。在KO样本中观察到较低的总分支长度与这些观察结果一致。在作为对照的两个KO样本之间,这些参数估计没有显著差异。相反,与WT相比,KO样本中的平均分支长度更高,因为KO神经突网络的密度较低,导致较少的交叉点,因此在较长的板段中。总的来说,这些结果表明,nHOTAIRM1对于spmn中神经突起的正常生长是必要的。
spMNs中受nHOTAIRM1缺失影响的其他基因包括参与“突触组织”和“化学突触传递调节”的基因。它们包括CACNA1D、CNR1、GRIA1、GRIK3和SHANK2基因,这些基因在KO spMNs中均显著下调(图7A)。
图7:WT和nHOTAIRM1有丝分裂后spmn。
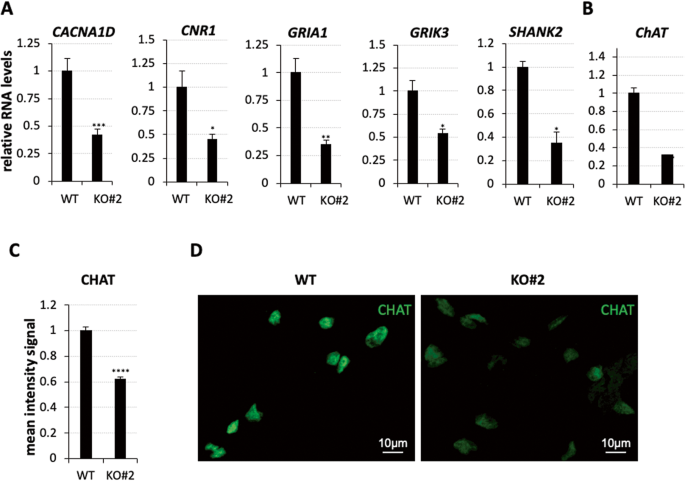
WT和nHOTAIRM1 KO (KO#2克隆)spmn (D12)突触基因的qRT-PCR分析(每个面板上显示)。数据(平均值±SEM)以任意单位表示,相对于atp50mrna水平,作为内部控制。对于每个目标,在WT spmn中的表达设为1。N=3个生物重复;* * * P≤0.05,P≤0.01,* * * P≤0.001,双尾学生的t检验。WT和nHOTAIRM1 KO (KO#2克隆)spmn (D12)中ChAT的qRT-PCR分析。数据(平均值±SEM)以任意单位表示,相对于atp50mrna水平,用作内部控制。在WT spmn中的表达设为1。N=3个生物重复;***P≤0.001,双尾Student 's t检验。C, D WT和nHOTAIRM1 KO (KO#2克隆)spmn免疫染色后CHAT蛋白强度信号的定量分析(D12)。对于每个样本,在两个不同的生物重复中随机选择260个细胞进行分析。**** p≤0.0001。非配对双尾学生t检验。
编码Cav1.3的CACNA1D基因的表达在KO spMNs中下降了约60%[40,41,42],而调节突触传递的CNR1基因的表达[43]下降了约56%。Cav1.3是电压门控L型Ca2+通道的成员,对突触成熟和修剪至关重要。在KO spmn中,负责MN树突结构的GRIA1[44]和与突触可塑性和增强相关的GRIK3[45]分别减少了约66%和47%。最后,影响突触连通性的SHANK2表达减少了约64%[46]。除了上述基因外,我们还关注了ChAT,这是一种编码胆碱乙酰转移酶的基因,该酶是合成乙酰胆碱的限速酶[47],对MNs的神经传递至关重要[48]。我们通过qRT-PCR(图7B)和IF(图7C, D)在D12 KO和WT spmn中评估其mRNA水平。我们发现,当nHOTAIRM1被耗尽时,它们的水平显著降低,这表明lncRNA在spmn中作为突触活动的调节剂发挥作用。
主要是细胞质,我们想知道nHOTAIRM1是如何发挥其靶基因的转录后控制的。细胞质lncrna的一个典型作用是竞争性内源RNA (ceRNA)参与microRNA海绵活性[49]。lnbase数据库(https://diana.e-ce.uth.gr/lncbasev3)未报道神经元系统中microRNA-nHOTAIRM1相互作用的数据。因此,我们研究了lncRNA与Argonaute 2 (AGO2)的相互作用,AGO2是mirna诱导沉默复合体(miRISC)的主要组成部分[50]。通过catRAPID算法(http://s.tartaglialab.com/page/catrapid_group)进行的计算机分析,预测任何给定转录物的蛋白伴侣,没有显示nHOTAIRM1对AGO2蛋白的结合倾向。因此,之前在spmn中鉴定其蛋白相互作用物的nHOTAIRM1 rap -质谱实验中没有检索到AGO2[12]。从分化神经元的细胞质提取物中进行的交联免疫沉淀(CLIP)试验进一步排除了nHOTAIRM1和AGO2之间直接相互作用的发生(补充图11)。总之,所有这些数据表明,nHOTAIRM1在被测试的神经元环境中确实具有作为ceRNA的先决条件。
另一种在编码rna和ncrna之间建立功能串扰的机制是由RNA-RNA相互作用介导的[49]。在天然条件下,对WT D12 spMNs的细胞质提取物进行RNA拉下实验。通过qRT-PCR分析富集nHOTAIRM1的下拉片段是否存在KO spmn中下调的mrna。在测试的候选转录本中,PROX1、ROBO1、SEMA6D、ChAT、GRIK3、CNR1、UNC5A和SHANK2在nHOTAIRM1下拉部分中显著富集(图8A)。为了检测活细胞中lncRNA-mRNA的直接配对,在spmn中进行了4 ' -氨基甲基-4,5 ',8-三甲基补骨脂素(AMT)交联RNA下拉试验。在原生RNA下拉实验中鉴定出的8个nHOTAIRM1 mRNA靶点中,有2个靶点,即ROBO1和SHANK2,被鉴定为lncRNA的直接相互作用物(图8B)。通过IntaRNA算法(http://rna.informatik.uni-freiburg.de/),我们检测了nHOTAIRM1与两个mrna之间的相互作用区域(Supplementary Fig. 12)。有趣的是,nHOTAIRM1建立RNA-RNA相互作用的区域包括外显子3的5 '端(核苷酸644-689),其中包括一个富含g的区域,根据QGRS制图工具(https://bioinformatics.ramapo.edu/QGRS/)预测,这是一个假定的g四重体形成序列(补充图13)。根据Ensembl (https://www.ensembl.org/)的研究,通过IntaRNA对ROBO1或SHANK2区域与nHOTAIRM1相互作用的计算预测表明,它们位于这两种mrna的5 '非翻译区(5 ' utr)。这一观察结果支持了nHOTAIRM1可能在mRNA稳定性和/或可译性控制中发挥作用的观点[51]。
图8:的识别nHOTAIRM1有丝分裂后spmn中的RNA相互作用物。
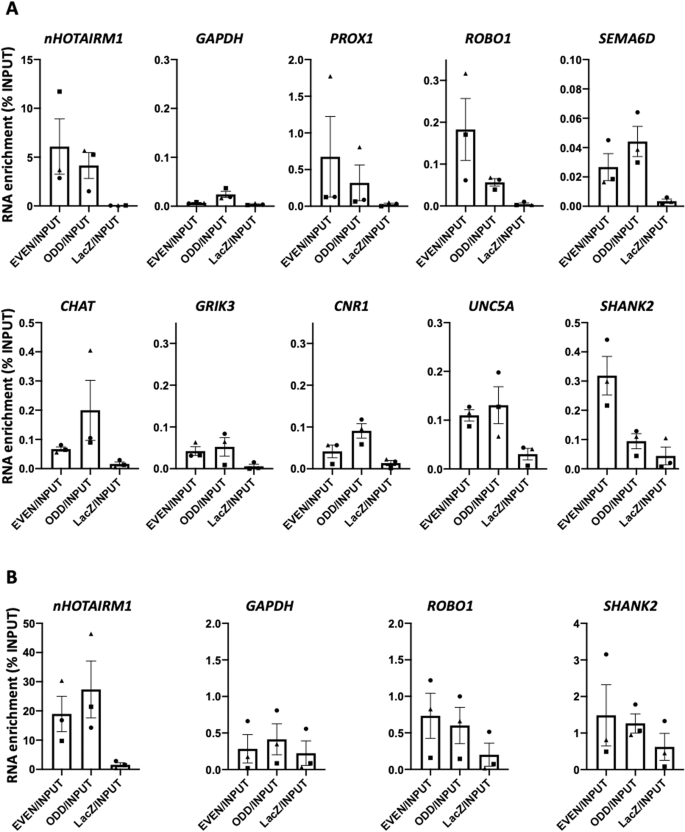
WT spMN (D12)细胞质提取物在自然条件下进行的nHOTAIRM1 RNA下拉实验的qRT-PCR分析。在WT spMN (D12)总提取物中进行的nHOTAIRM1 amt交联RNA下拉实验的qRT-PCR分析。在两个面板中,数据(平均值±SEM)以相对于输入的RNA富集百分比表示。箱形图表示N=3个独立生物重复的平均值。符号(三角形、正方形和圆形)表示每个生物复制的值。
总的来说,这些结果强调了lncRNA-mRNA串扰在导致功能性spmn产生的多步骤过程中的影响。
摘要。
介绍
结果
讨论
材料与方法
数据可用性
参考文献。
致谢。
作者信息
道德声明
补充信息
# # # # #
这项研究表明,lncRNA nHOTAIRM1在调控spMN产生和功能的调控网络中是一个新的参与者。由于在SC中特异性表达,我们探索了它在人类ipsc衍生的spmn中的功能,它位于体细胞和神经突中(图1C, D)。应用反向遗传学方法结合转录组学分析、RNA下拉实验和RNA FISH/IF分析,我们发现了nHOTAIRM1的广谱调控作用。
在功能性spMN中鉴定出nHOTAIRM1靶基因,当lncRNA缺失时,其表达显著改变,证明nHOTAIRM1参与(i)以in命运为代价的spMN分化,(ii)神经突生长,以及(iii)突触传递的调节。
值得注意的是,nHOTAIRM1的缺失导致多个分化基因的强烈下调,其中HB9及其调控因子OLIG2是触发MN规范和抑制IN承诺的关键。因此,在iPSCs转化为spmn的过程中,lncRNA的缺失会导致特定MN标记物的急剧减少,如ChAT[30]和ISLET1[参考文献[4]](图5A),并伴随着spmn数量的减少,其标志是ChAT和ISLET1蛋白的共表达(图5B)。
与HB9在小鼠诱导的MN中不起作用时所发生的情况类似[31],由此推论,nhotairm1依赖性MN标记的减少伴随着特异性in标记的诱导(图5C和补充图9),这表明lncRNA作为MN前分子,在层次上位于OLIG2/HB9模块的上游,处于MN和in细胞命运的交叉位置(补充图9B)。
为了将nHOTAIRM1更全面地纳入神经元分化的复杂调控网络,还应考虑其在控制神经元前体和分化神经元之间转换中的作用。事实上,我们证明lncRNA是抑制主前神经TF神经原素2 (NGN2)及其下游神经原性通路的表达所必需的[12],从而使分化得以进行,并维持获得性神经元细胞的身份。这个调节轴与OLIG2和NGN2在SC发育中所起的作用相吻合。在MNPs中,OLIG2表达通过抑制有丝分裂后MN基因(如HB9)来保持其未分化状态。另一方面,OLIG2在一部分祖细胞中触发NGN2的表达,使它们成为MNs。因此,OLIG2和NGN2的相对水平控制着将MN祖细胞转化为有丝分裂后MN的细胞命运决定[29]。nHOTAIRM1同时作用于OLIG2和NGN2,可能有助于确定它们的适当比例。由此产生的整体图像与nHOTAIRM1位于由OLIG2、NGN2及其靶标组成的复杂分子电路的顶端的推测是一致的,它在分化的SC中充当计时正确(运动)神经元基因表达的大门。
关于nHOTAIRM1参与建立适当的神经元连接,我们在分子和表型水平上对其进行了评估。nHOTAIRM1的缺失导致影响神经元连接和靶向(NrCAM)、轴突引导(SEMA3E和SEMA6D)以及SC外运动轴突轨迹(DCC和ROBO1)的基因显著下调(图6A)。表型上,nHOTAIRM1的缺失导致MN形态缺陷(图6B),分支和连接数量以及总分支长度显著减少(图6D-F)。
与此密切相关的是nHOTAIRM1对编码突触活动调节剂的靶基因的控制(图7A, B),其功能障碍与中枢神经系统疾病有关。这表明nHOTAIRM1在spmn中的病理生理作用。例如,在肌萎缩侧索硬化症(ALS)患者的SC中观察到ChAT活性降低,这被认为与MN功能丧失有关[52](图7B, C)。根据这一假设,我们证明了nHOTAIRM1与RNA结合蛋白FUS的物理相互作用,nHOTAIRM1的表达取决于FUS的水平[12],FUS的功能丧失会导致神经元功能障碍和ALS神经退行性变[53]。基于这一证据,我们计划在未来更好地揭示nHOTAIRM1与ALS突变体FUS之间的相互作用。
从机制上讲,nHOTAIRM1与许多靶基因建立了RNA-RNA相互作用。PROX1、SEMA6D、ChAT、GRIK3、CNR1和UNC5A mrna与nHOTAIRM1建立间接相互作用(图8A),而ROBO1和SHANK2 mrna直接与lncRNA相互作用(图8B)。我们很容易推测直接和间接靶点与lncRNA的动态关联可能在spMN分化过程中发挥作用。由于这些相互作用在有丝分裂后mn的细胞质中被检测到,我们提出nHOTAIRM1作为spMN基因表达的转录后调节因子。据我们所知,这是人类spmn中lncrna介导的转录后控制的第一个证据,之前在小鼠MNs中描述过的唯一一个例子[54]。预测nHOTAIRM1, ROBO1和SHANK2的直接mRNA相互作用物在其5 ' utr上结合,需要进一步的机制研究来阐明lncRNA是否在其稳定性或翻译水平上调节其靶mRNA。有趣的是,RNA-RNA相互作用预测也表明nHOTAIRM1结合可能通过假定的rG4结构发生。已知rG4s参与基因表达、lncRNA功能和疾病进展的调节[55]。
总之,我们的研究结果表明,nHOTAIRM1作为参与MN分化、树杈化和活性的多种分子途径的多效调节因子,对于确保功能性spmn的产生至关重要。由于其活性影响SC细胞的命运决定,该研究也为未来研究阐明nHOTAIRM1如何在负责运动整体协调的运动回路中促进神经元亚型之间的正确平衡提供了理论基础。
本研究中使用的人诱导多能干细胞(iPSC-NIL)是按照文献[14]中描述的方法获得、维持并诱导分化为spMNs的。
使用Benchling设计工具(https://www.benchling.com/)设计针对HOTAIRM1位点区域的sgrna(数据集2)。结合Zenbu (https://fantom.gsc.riken.jp/zenbu/)的FANTOM5转录起始位点数据,选择Cas9双链断裂靶区。编码WT Cas9蛋白的PX333质粒从Addgene购买,sgRNAs作为单链DNA探针(Bio-Fab Research - Rome, IT)订购,并按照Zhang Lab Protocol (https://media.addgene.org/data/plasmids/62/62987/62987attachment_KiOWQSPn2egB.pdf)的建议进行克隆,得到PX333-sgRNAs载体。HR110PA (System Biosciences - Palo Alto, CA, USA)作为主干创建供体载体(donor)。使用In-Fusion?HD Cloning Plus Kit (Cat.)将Poly-adenylation序列(PAS)克隆到供体中,随后是新霉素耐药盒。# 638910)。利用iPSC-NIL gDNA (Kapa HiFi, Takara Bio - Saint-Germain-en-Laye, FR), PCR扩增出长度为800 nt的两条同源臂(HA) HA1和HA2。HA1克隆在PAS的上游,HA2克隆在PAS的下游。通过Neon转染系统(Life Technologies - Carlsbad, CA, USA)在基质包被的培养皿上转染iPSCs,使用100 μl尖端在R缓冲液中,设置为:1200 V, 30 ms, 1脉冲。用800 μg/ml G418进行筛选,连续5 d。扩增HOTAIRM1 KO单克隆并进行基因分型。
利用Ambion PARIS试剂盒(AM1921, Life Technologies)对ipsc衍生的spmn进行亚细胞分离。提取RNA后,逆转录等量的细胞质和细胞核RNA,并进行qRT-PCR分析。归一化是基于RNA的总量。
从ipsc衍生的spMNs中使用改良Boyden室进行体细胞/神经突分离,并通过TUBB3蛋白免疫染色进行评估,如[19]所示。然后用qRT-PCR分析spMN体细胞和神经突的RNA样本。神经突室中神经元投射标记物COL3A1和体细胞中GNG3的富集可作为两个室适当分离的对照。
使用TruSeq搁浅mRNA文库准备试剂盒(Illumina - San Diego, CA, USA)从ipsc衍生的WT和nHOTAIRM1 KO spmn中提取的polyA+ RNA(3个独立的生物重复)获得测序文库。
测序反应产生100个核苷酸长的对端reads,在Novaseq 6000测序系统(Illumina)上进行,深度超过20 M。去除转接序列和低质量端基使用Trim Galore188 (version 0.6.4_dev)软件;修剪后的最小读取长度设置为20。使用STAR(版本2.7.9a189)对人类GRCh38基因组初级组装进行比对。
大约85%的总读数被成功地映射到人类基因组,其中大多数与独特的位置对齐。使用quantMode转录esam选项生成翻译成转录物坐标的比对。采用RSEM方法定量转录本的表达水平。
使用Bioconductor软件包DESeq2进行差异表达分析,该软件包符合每个基因的负二项广义线性模型。只有在至少三个样本中显示最小绝对表达值为10的基因(如DESeq2包开发人员所建议的)被保留。收缩日志折叠变化值是由lfcShrink函数获得的,该函数考虑不是由于低计数引起的最大折叠变化,并使用这些变化来生成先验分布。padj < 0.05和绝对logFC > 1的基因被认为是差异表达,并用于进一步分析。
使用GO数据库(http://geneontology.org/)上的WebGestalt R工具,对padj < 0.05和绝对logFC > 1的显著上调和下调基因进行GO过度代表性分析(ORA)。
使用IntaRNA版本3.3.2178绘制amt交联RNA下拉实验中发现的nHOTAIRM1与mRNA相互作用物之间的结合区域。为了获得RNA-RNA相互作用的计算机预测,我们通过观察RSEM输出异构体,从RNA- seq结果中过滤出在我们的系统中未表达的RNA转录异构体。
在表达的剪接异构体中,我们考虑了WT和KO样品中的TPM表达值,以选择在我们的系统中高表达的蛋白质编码剪接异构体。随后,我们检索了这些转录本的FASTA文件,并使用带有默认参数的IntaRNA来预测nHOTAIRM1的mRNA靶位点。
总RNA采用Direct-zol RNA MiniPrep (R2052, zimo Research - Irvine, CA, USA)提取。qRT-PCR检测采用Takara Primescript RT Reagent Kit (RR037A, Takara Bio)合成cDNA。使用SensiFAST SYBR Lo-ROX试剂盒(BIO-94020, Bioline - Memphis, TN, USA)在7500 Fast Real-Time PCR (Applied Biosystem - Carlsbad, CA, USA)上进行qRT-PCR检测。以atp50mrna作为参考靶标。RNA下拉实验中,使用miRNeasy Micro Kit (Qiagen - Germantown, MD, USA)提取总RNA,使用Superscript VILO cDNA Synthesis Kit (ThermoFisher Scientific - Carlsbad, CA, USA)合成cDNA。
ipsc衍生的MNs被镀在12 mm直径的盖上,盖上涂有基质(hesc合格的Matrix Corning 354277),并在4%多聚甲醛中固定(Electron Microscopy Sciences - Hatfield, PA, USA),在室温下固定10分钟。脱水步骤用冷冻乙醇系列(50%,70%,100%)进行,以便在- 20°C保存细胞直到使用。
如文献[56,57]所述,nHOTAIRM1通过RNA FISH使用18种生物素化探针(数据集2)进行检测。MNs通过低温乙醇系列(100%,70%,50%)再水化,并在含有0.05% Triton X-100和2 mM VRC (R3380, Sigma-Aldrich - St. Louis, MO, USA)的DPBS溶液中渗透5分钟。然后将细胞在DPBS中洗涤三次,然后用2X SSC缓冲液(3 M NaCl;0,3 M柠檬酸钠在无核酸酶的水为20倍的原液)。SSC孵育5分钟后,用预杂交缓冲液孵育(10%去离子化甲酰胺,47671,Sigma-Aldrich,;2X SSC(无核酸酶水)在37°C下保存15分钟。然后在载玻片杂交机(ACD HybEZ?II杂交系统)中37°C孵育MNs,杂交缓冲液(10%去离子化的乙酰胺;2 x SSC;10% w/v硫酸葡聚糖,2 mM VRC在无核酸酶的水中)补充生物素化探针,每个最终浓度为50 nM。第二天,细胞用2X SSC在37℃和室温下洗涤两次,洗涤5分钟,然后丢弃SSC缓冲液,用TN缓冲液(Tris HCl pH 7.5 1 M;NaCl 5 M(无核酸酶的水),在室温下作用10分钟。最后,生物素化寡核苷酸用1:200稀释的568-共轭链亲和素(S11226, Invitrogen - Waltham, MO, USA,)在4% w/v BSA/TN缓冲液中在潮湿箱中RT下孵育1-2小时。
FISH与IF联合染色时,细胞用TN缓冲液洗涤2次,用DPBS洗涤1次,RT下孵育5min,然后用一抗(抗β tubiii, T2200, Sigma Aldrich;anti-Map2, 17490-1-AP, Proteintech, Manchester, UK)用1% w/v BSA/DPBS稀释。随后,用DPBS冲洗样品3次,RT下5分钟,RT下用二抗(Goat anti-Mouse 488, A-11029, Invitrogen;山羊抗兔488,A-11008, Invitrogen;驴抗兔594,IS-20152-1,免疫学科学-罗马,IT)在1% w/v BSA/DPBS中稀释1:300。最后,用DPBS冲洗细胞3次,RT下5分钟,细胞核用DAPI溶液(1ug/ml/PBS, D9542, Sigma Aldrich)反染,RT下5分钟,盖片用延长钻石安装介质(P-36961, ThermoFisher Scientific)安装。
在0.01% poly-L-ornithine/Murine Laminin 20 μg/ml (Sigma Aldrich)培养基中培养D12的MNs,在4%多聚甲醛(Electron Microscopy Sciences)培养基中4℃固定20分钟,PBS洗涤。然后用0.2% Triton X-100/3% BSA/PBS对固定细胞进行渗透和阻断,在室温下孵育15分钟。随后,用抗chat (ab223346 - Abcam, Cambridge, UK)和抗islet1 /2一抗(小鼠克隆39.4D5 - DSHB, Iowa City, IA, USA)分别在3% BSA/PBS溶液中稀释1/200和1/50,在4°C下孵育细胞。PBS洗涤后,细胞在含有1%驴血清、1%山羊血清PBS、驴抗小鼠647 1:30 00 (A32787, Invitrogen)和山羊抗兔555 1:30 00 (A32732, Invitrogen)的溶液中RT孵育45分钟。细胞核用DAPI溶液(1μg/ml/PBS, D9542, Sigma Aldrich)反染。
图像采集采用倒置共聚焦Olympus IX73显微镜,配备Crestoptics X-LIGHT V3旋转磁盘系统和Prime BSI Express Scientific CMOS相机,UPlanSApo 60X (NA 1.42)油物镜,并使用metaMorph软件(Molecular Devices - San Jose, CA, USA)收集。自动拍摄z -叠共聚焦显微镜图像(z -间距200nm)。
使用ImageJ软件对CHAT的信号强度进行最大强度投影分析,并对每个单个细胞进行测量。在两个不同的生物重复中随机选择260个细胞进行WT和KO实验。使用“Analyze Particle”工具对ImageJ上自动阈值获取的二值图像进行CHAT + /ISLET1+细胞计数。
神经突网络分析在ipsc衍生的WT、nHOTAIRM1 KO#1和KO#2 spmn中进行,如[39]所述,并进行了轻微修改。根据文献[39]中描述的工作流程,创建了用于MN神经突网络半自动分析的Fiji-imageJ宏。简单地说,用神经突标记物Map2染色WT和nHOTAIRM1 KO spmn共聚焦图像生成z投影[参考文献][58,59,60]]。为了从DAPI通道开始分割体细胞,使用Huang阈值算法生成细胞核掩模,并去除半径小于20 px的异常值。利用ImageJ的“放大选择”功能,从细胞核掩膜开始生成一个选择,然后将其扩大5 px (1 px=0.207 mm),以解释成熟细胞核典型的小细胞质部分。
随后,复制Map2通道并生成两个不同的掩码。从第一个副本开始,通过对比度增强(饱和pxs=0.1%)和高斯模糊(σ=2 μm)滤波,使用Moments算法创建高强度蒙版阈值。这个掩模覆盖了图像的高强度部分,包括体细胞、轴突丘和神经突边缘。第二个副本用于生成低强度掩模,LoG掩模,以解释神经突较薄的部分。为了创建LoG掩码,对比度增强(饱和pxs=0.1%),使用Process > Math ImageJ菜单中的LoG过滤器,并使用Moments算法对图像应用阈值。
然后使用ImageJ的“Image Calculator”功能将高强度掩码与LoG掩码合并,再减去核掩码,得到最终的神经突掩码。
最后,对神经突掩膜进行骨架化处理,利用“分析骨架”功能确定每幅图像的分支、结点和端点数量,以及总分支长度和平均分支长度。
利用多点工具,从细胞核掩模开始手动计算每张图像的细胞数量。样品在配备1.49 NA ×100物镜(Apo TIRF 100x Oil, Nikon, Tokyo, Japan)和60倍物镜的Nikon Instrument A1共聚焦激光显微镜上成像。使用NIS-Elements AR软件(Nikon, Tokyo, JP)采集共聚焦图像,使用ND采集模块对厚度为4 mm的z堆叠图像(z间距150-175 nm)进行多点采集。
如文献[61]所述,对ipsc衍生的spMNs (D12)的细胞质提取物进行天然RNA下拉实验。用于提取nHOTAIRM1的生物素化DNA探针列在数据集2中。
amt交联RNA下拉实验如[62]所述。用于提取nHOTAIRM1的生物素化DNA探针列在数据集2中。
CLIP实验在本研究中描述的经ra处理的SH-SY5Y神经母细胞瘤细胞的细胞质提取物中进行[12]。
直方图显示了至少三个独立生物重复的平均值±SEM。N在图图例中表示。误差是从相对数量计算出来的,然后适当地传播;采用双尾配对或非配对Student’s t检验确定统计学显著性,见图图例。p值< 0.05认为有统计学意义。* * * P < 0.05, P < 0.01, * * * * * * P < 0.001, P < 0.0001。
ccDownload: /内容/ pdf / 10.1038 / s41419 - 023 - 06196 - y.pdf

