

衰老时钟的建立强调了DNA甲基化变化与衰老之间的紧密联系。然而,目前尚不清楚是否可以使用其他表观遗传特征来准确预测年龄。此外,先前的研究已经观察到缺乏与年龄相关的DNA甲基化变化对基因表达的影响,这使得基于DNA甲基化的衰老时钟的可解释性受到质疑。在这项研究中,我们探索利用染色质可及性来构建衰老时钟。我们收集了159名献血者的血液,并生成了染色质可及性、转录组学和细胞组成数据。我们研究了衰老过程中染色质可及性的变化,构建了一个中位数绝对误差为5.27年的新型衰老时钟。时钟使用的染色质可及性的变化与转录组学改变密切相关,有助于时钟解释。我们还表明,我们的染色质可及性时钟比匹配样本上训练的转录组时钟表现得更好。总之,我们证明生物钟依赖于细胞内在染色质可及性的改变,而不是细胞组成的变化。此外,我们提出了一种基于染色质可及性构建表观遗传衰老时钟的新方法,该方法与年龄相关的转录改变有直接联系,但比转录组时钟可以更准确地预测年龄。
衰老是一个生物学过程,其特征是在多个生物学尺度上逐渐丧失生理完整性,对疾病和死亡的脆弱性增加[1]。当前全球人口老龄化趋势突出了研究老龄化的重要性,以了解其动态并减轻其作为晚年疾病驱动因素的作用[2]。
表观遗传扰动是衰老的一个标志[1,3]。特别是,基于dna甲基化的衰老时钟的发展表明,甲基化的变化发生在整个生命过程中,可以用来准确预测年龄[4,5,6,7,8,9]。自从甲基化时钟的发现以来,该领域已经表明时钟可以从其他生物信号中构建,例如转录组或蛋白质组谱[10,11,12,13,14,15,16,17]。尽管探索了这些不同的信号,表观遗传时钟迄今为止只使用DNA甲基化,部分原因是数据的可用性。尽管如此,表观遗传调控包括DNA甲基化以外的许多机制[3]。这些不同层次的表观遗传调控是否可以用来预测年龄仍然是一个悬而未决的问题。此外,由于单个CpGs的甲基化与下游基因的转录相关性较差,因此很难解释哪些生物过程对应于时钟使用的甲基化特征[18]。基于这些原因,我们试图创建一个基于染色质可及性的衰老时钟。染色质可及性整合了多种表观遗传机制的影响,因此提供了比DNA甲基化更全面的染色质状态描述[19]。先前的研究已经观察到多种生物中染色质可及性的年龄相关变化[20,21,22,23]。此外,衰老的异染色质损失理论源于对衰老过程中染色质整体去抑制的观察[24,25]。我们设想,这些与年龄相关的变化将允许构建时钟,并扩大我们对表观遗传失调的理解。
在这项研究中,我们分别使用ATAC-seq[26,27]和RNA-seq从广泛年龄范围的人类血液样本中生成了染色质可及性和转录组学图谱。然后,我们分析了与年龄相关的可及性变化以及它们与转录组的关系。随后,我们使用弹性网络回归模型从染色质可接近性剖面预测年龄,并具有良好的准确性。最后,我们通过研究其预测因子并将其性能与基于转录组的时钟进行比较来表征时钟。
从159名健康献血者(117名男性,42名女性)获得血液样本,年龄从20岁到74岁不等(图1a)。从157个样本中分离外周血单个核细胞(PBMCs)以生成ATAC-seq图谱,其中143个(105个男性,38个女性)通过了质量控制(年龄和性别分布包括在补充图1a中)。片段大小分布的代表性直方图见补充图2b)。从这些样本中,我们共检测到80,400个开放染色质区域(ocr),其中24.1%位于转录起始位点(TSS) 1 kbp内,因此被注释为启动子,58.2%包含具有增强子活性的位点,5.0%被注释为启动子和增强子。其余12.7%的ocr不位于tss附近,没有报告增强子活性,将被称为“未注释”(图1c)。可达性剖面的主成分分析将样本置于沿PC1的老化轨迹上(PC1-age Pearson’s r=0.35, p=2.14e-5,补充图1c)。此外,我们对所有159个样本进行了RNA-seq,从中我们检测到16155个基因的表达。在这些样本中,有144个样本通过了质量控制(年龄和性别分布见补充图1b), 132个样本有匹配的ATAC-seq样本,也通过了质量控制。表达谱的主成分分析将样本置于沿PC1和pc2的老化轨迹上(PC1-age Pearson 's r=-0.27, p=9.89e-4, PC2-age Pearson 's r=0.26, p=1.86e-3,补充图1c)。最后,我们用流式细胞术测量了所有样品中单核细胞、粒细胞、淋巴细胞、总T细胞、CD4 + T细胞、CD8 + T细胞、B细胞和NK细胞的比例(补充图2a,补充图3a-g)。在衰老过程中,我们检测到NK细胞比例增加(Pearson’s r=0.31, p=1e-4),总T细胞数量减少(Pearson’s r=-0.22, p=5.3e-3)和CD8 + T细胞数量减少(Pearson’s r=-0.24, p=2.4e-3)。单核细胞、粒细胞、淋巴细胞、CD4 + T细胞和B细胞的比例与年龄无显著相关。在先前的研究中也报道了PBMC成分的类似变化[22,28,29]。
图1
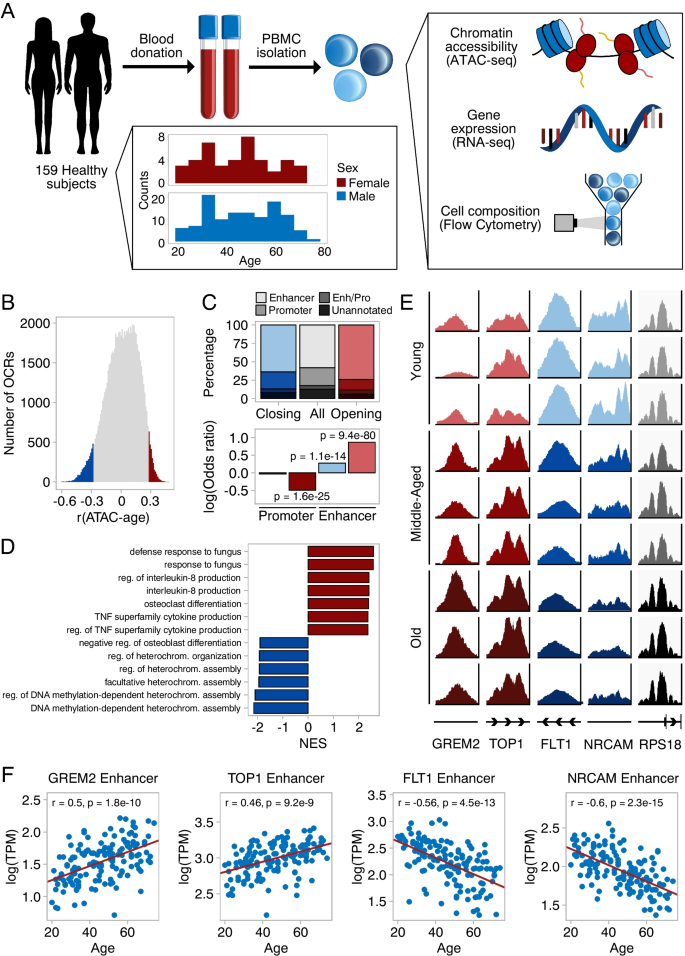
在衰老过程中染色质可接近性发生变化。(A)从年龄分布广泛(20-74岁)的159名健康献血者的血液样本中分离出pbmc。对所有样品进行ATAC-seq、RNA-seq和流式细胞术分析。(B)染色质可及性与年龄之间的相关性分布(Spearman’s r)。具有统计学意义的闭合ocr以蓝色突出,具有统计学意义的打开ocr以红色突出(FDR < 0.01)。(C)统计上显著的ocr对调控元件的注释。开启和关闭ocr间启动子和增强子的富集。Log(优势比)和p值采用Fisher 's Exact检验计算。(D)衰老过程中染色质可及性的GSEA变化。根据归一化富集分数(NES)绘制基因本体生物过程项。显示了前六个正(红色)和负(蓝色)NES的术语。(E)不同年龄的代表性样本的前两个开放和关闭ocr的可达性概况。y轴使用用于原始计数归一化的相同比例因子重新缩放。最后一列表示在衰老过程中其可及性没有改变的持家基因。年轻:20-22岁;中年:45-47岁,老年:70-71岁。(F)年龄相关性最强的4个ocr的染色质可及性(log(TPM))随年龄的散点图。Pearson的r值和p值均已显示
为了了解衰老对表观基因组的影响,我们分析了染色质可及性的全局和位点特异性变化。根据衰老的异染色质损失理论,我们可能会看到ocr外染色质的逐渐去抑制。随着年龄的增长,ocr内读数的比例呈负趋势,但不显著(Pearson’s r=-0.13, p=0.12, Supplementary Fig. 4a)。然而,我们确实注意到,ocr内读取的部分对技术变化很敏感。因此,我们询问是否可以通过其他手段观察全局变化,例如启动子处OCR宽度的变化或重复元件的去抑制。在年轻供体(< 35岁,n=40)和老年供体(> 55岁,n=44)之间,转录起始位点(TSS)周围的平均覆盖情况没有差异(补充图4c)。同样,我们也没有看到重复元件的可及性在总体上(Pearson’s r=0.13, p=0.14, Supplementary Fig. 4b, d)以及单独考虑重复元件家族时(Supplementary Fig. 4e)的显著增加。
至于位点特异性变化,我们在2622个ocr中观察到染色质随年龄的开放,在3765个ocr中观察到染色质随年龄的关闭(Spearman’s r, FDR < 0.01,图1b)。图1e显示了几个ocr的覆盖概况的例子,这些ocr的打开、关闭或不随年龄变化,而图1f显示了相同ocr的可及性与年龄之间的相关性。在开放的ocr中,6.0%被标注为启动子和增强子,14.3%为启动子,74.0%为增强子,5.8%未标注。在结束的ocr中,5.4%被标注为启动子和增强子,23.1%为启动子,63.7%为增强子,7.8%未标注。有趣的是,我们观察到与背景相比,打开和关闭ocr中的增强子都显著富集(Fisher精确检验打开p=9.44e-80,关闭p=1.08e-14,图1c)。相反,启动子在启动ocr中被耗尽,而在关闭ocr中没有(Fisher 's精确检验p=1.6e-25, p=0.38)。这表明增强剂可能对衰老过程中可及性的变化特别敏感。接下来,我们将ocr与基因联系起来,并研究了它们在生物过程中的作用。我们将ocr与其最近的基因联系起来,并进行了GSEA[30](图1d,补充文件4)。富集分数为正的项包括IL8和TNF的产生以及对真菌的防御,而富集分数为负的项则与异染色质组装的调节有关,包括依赖于DNA甲基化的异染色质组装。此外,我们仅对启动子进行GSEA(补充图5a),或使用PEREGRINE数据集将增强子与下游基因关联起来:PEREGRINE数据集是基于包括血液在内的多种组织的china - pet、eQTL和Hi-C预测的增强子基因链接集合[31](补充图5b)。与之前的方法一样,具有正富集分数的术语与炎症有关,而具有负富集分数的术语与染色质组装的调节有关。
总之,我们发现pbmc的染色质可及性在衰老过程中没有显著的全局变化,至少在我们分析的年龄范围内(20-74岁)。相反,我们检测到特定调控元件的变化,最常见的是增强子,它与炎症增加和异染色质组装减少有关。尤其令人困惑的是,在参与异染色质组装的上游基因中,发现与年龄相关的ocr抑制,而没有观察到显著的全球去抑制。值得一提的是,ATAC-seq只能提供可及性的相对量化,因此,可能无法检测到全基因组范围内可及性的均匀增加。尽管如此,特定ocr的可及性与年龄之间的相关性表明,确实有可能构建一个基于染色质可及性的衰老时钟,类似于DNA甲基化所做的。
基于DNA甲基化的衰老时钟的一个局限性是,甲基化的变化很难与下游细胞过程联系起来,从而限制了它们的可解释性。先前的研究发现,在年龄阶段,差异甲基化区下游基因的转录通常不随甲基化而变化[18]。同一项研究发现,超甲基化主要影响那些表达本来就很低的基因,从而解释了对转录明显缺乏影响的原因。
因此,我们想知道在我们的数据集中,我们是否能够观察到与染色质可及性变化一致的表达变化。在16,155个表达基因中,随着年龄的增长,440个表达增加,544个表达减少(Spearman’s r, FDR < 0.01, Supplementary Fig. 5c)。GSEA中富集分数为阳性的术语(图2a,补充文件7)与病原体反应(对细菌来源分子的反应,对脂多糖的反应)和凝血(止血,凝血)有关。负富集评分与B细胞活性和补体激活(B细胞受体信号通路,补体激活,循环Ig的体液反应)相关的术语。这些术语表明免疫功能和炎症的改变,与我们看到的染色质可及性一致。与异染色质组装相关的术语也在我们的转录组学数据中丰富,但程度低于染色质可及性。通过更详细的调查,我们确定了几个基因,它们的表达和在调控元件上的可及性都与年龄相关(图2b, h显示了一个这样的基因:CD248的覆盖图),并试图确定这些基因是否比偶然预期的更常见。因此,我们比较了与ocr相关基因的年龄相关性,与年龄呈正相关(Spearman r > 0, FDR < 0.01),与年龄负相关(Spearman r < 0, FDR < 0.01),与年龄不相关(FDR > 0.01)。我们发现,总体而言,与可及性随年龄增长而增加的启动子相关的基因在衰老过程中表达上调(单侧Kolmogorov-Smirnov检验,D=0.33, p < 0.001,图2c),类似地,与年龄关闭的启动子相关的基因在衰老过程中倾向于下调(单侧Kolmogorov-Smirnov检验,D=0.34, p < 0.001,图2c)。当我们观察可达性随年龄增长而增加的增强子(单侧Kolmogorov-Smirnov检验,D=0.15, p < 0.001,图2c)和可达性随年龄增长而下降的增强子(单侧Kolmogorov-Smirnov检验,D=0.25, p < 0.001,图2c)时,这种模式较弱,但仍然非常显著。我们使用PEREGRINE基因增强子链接重复了这一分析,但发现这样做降低了染色质可及性和转录组学数据之间的一致性(补充图5d)。即使只考虑在血液中验证的基因增强子链接,这种一致性也没有得到改善(补充图5d)。最后,我们想知道与甲基化水平和转录之间的关联相比,可及性和转录之间的关系的强度如何。因此,我们使用Hannum等人[6]的公开DNA甲基化数据集来计算甲基化与年龄的相关性(Spearman’s r),并评估全基因组范围内转录、可及性和甲基化的年龄相关性之间的配对相关性(图2f, g)。我们发现,在增强子和启动子中,甲基化的年龄相关变化与转录组改变几乎没有相关性(Pearson’s r分别=-0.017和-0.051)。相比之下,可及性的变化与转录的变化相关,尤其是启动子的变化(启动子的Pearson’s r=0.318,增强子的r=0.252)。有趣的是,可及性的变化与增强子的甲基化变化相关(r=-0.198),而与启动子的甲基化变化相关较弱(r=-0.045)。我们还重复了这一比较,重点是甲基化水平与年龄显著相关的CpGs (Spearman’s r, FDR < 0.01,图2d, e),以确保我们不会错过非线性关系。在启动子和增强子上,CpGs低甲基化下游基因的年龄相关性显著向正值转移,尽管效应大小很小(单尾Kolmogorov-Smirnov检验,启动子的D=0.022, p=4.3e-6,增强子的D=0.055, p=4.7e-11)。在启动子位置的CpGs超甲基化下游基因(D=0.016, p=2.8e-4)则相反(D=0.020, p=0.077),而在增强子位置则不然。因此,即使关注在衰老过程中甲基化发生极端变化的CpGs,我们也没有看到对转录的影响。
图2
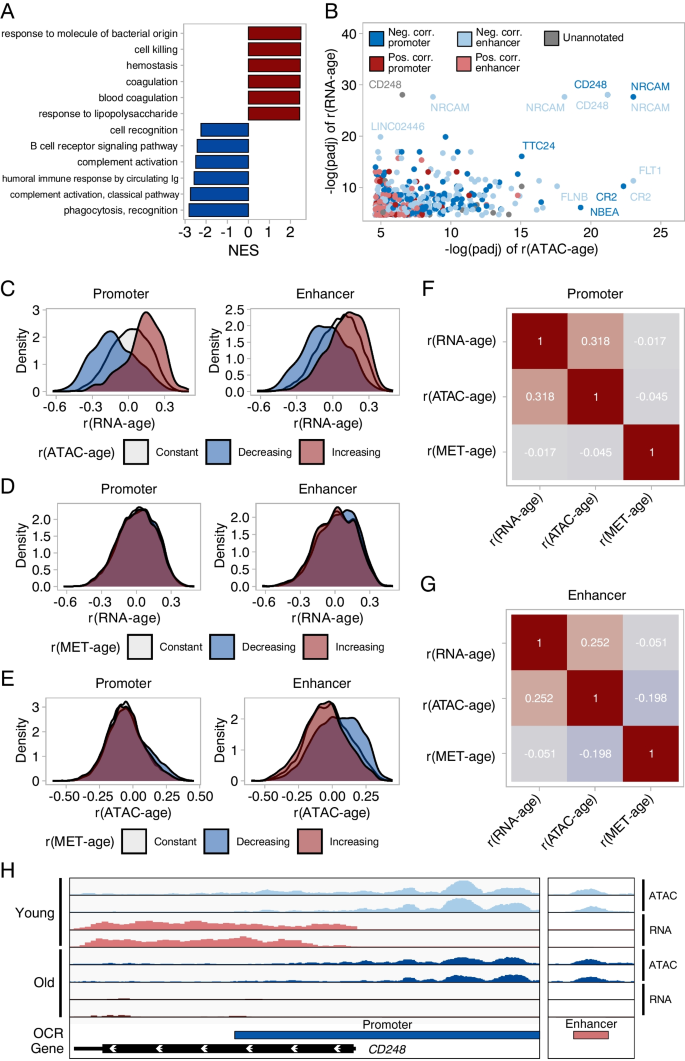
衰老过程中基因表达、染色质可及性和DNA甲基化的综合分析。(A) GSEA基因在衰老过程中的表达变化。根据归一化富集分数(NES)绘制基因本体生物过程项。显示了前六个正(红色)和负(蓝色)NES的术语。(B)基因在调控元件上的表达和可及性都与年龄相关(Spearman’s r)。x轴表示ATAC-seq数据中相关的显著性,y轴表示RNA-seq数据中相关的显著性。相关性的显著性由经fdr校正的p值的-log表示。(C)启动子/增强子相关基因的年龄相关性分布,在衰老过程中打开(Spearman’s r > 0, FDR < 0.01),在衰老过程中关闭(Spearman’s r < 0, FDR < 0.01)或不改变(FDR≥0.01)。(D)在衰老过程中获得甲基化(Spearman’s r > 0, FDR < 0.01),在衰老过程中失去甲基化(Spearman’s r < 0, FDR < 0.01)或不改变(FDR≥0.01)的启动子/增强子中CpGs相关基因的表达年龄相关性分布。(E) CpGs在衰老过程中甲基化增加(Spearman’s r > 0, FDR < 0.01)、甲基化减少(Spearman’s r < 0, FDR < 0.01)或不变化(FDR≥0.01)的ocr可及性年龄相关性分布。(F)基因表达年龄相关性、可及性年龄相关性、CpG甲基化年龄相关性之间的两两相关性,特别是在启动子区域。(G)基因表达年龄相关性、可及性年龄相关性、CpG甲基化年龄相关性之间的两两相关,特别是在增强子区域。(H)基因CD248的ATAC-seq和RNA-seq覆盖轨迹,其启动子和增强子的表达和可及性都随着年龄的增长而下降。展示了两个年轻的和两个年老的样品。y轴使用用于原始计数归一化的相同比例因子重新缩放
然而,重要的是要考虑到,在这种比较中,甲基化和转录之间的关联强度可能被低估,因为甲基化和表达数据没有在匹配的样本中产生。为了进行更公平的比较,我们将我们的RNA-seq数据替换为Marquez等人的另一个PBMC RNA-seq数据集[22]。令我们惊讶的是,年龄相关的染色质可及性变化仍然与转录组改变密切相关(补充图5e, f,启动子的皮尔逊r=0.314,增强子的r=0.235),而甲基化变化与表达变化之间的相关性仍然很弱(启动子的皮尔逊r=-0.023,增强子的r=-0.046)。最后,先前的报道发现基因表达与基因体甲基化的关系可能不同于基因表达与启动子/增强子甲基化的关系[32]。因此,我们将年龄相关的基因体甲基化变化与年龄相关的基因表达变化联系起来,但再次发现相关性非常弱(使用我们的RNA-seq数据时Pearson的r=-0.034,使用Marquez等人的数据时r=-0.026)。两种情况均使用Hannum等人的甲基化数据)。
因此,我们发现衰老过程中染色质可及性的变化与连贯的转录改变有关。另一方面,甲基化变化与年龄相关的表达变化的相关性非常弱。因此,我们得出结论,构建在染色质可及性上的时钟将与转录组改变及其对生物过程的影响直接相关。
随着年龄的增长,我们发现了染色质可及性的许多位点特异性变化,我们研究了这些变化是否可以用来预测献血者的年龄。为此,我们对通过质量控制的143个ATAC-seq样本进行了弹性网络回归模型的训练。我们使用嵌套交叉验证来调整超参数并估计模型的性能。在巢式交叉验证的外折叠中,模型选择183±58个ocr作为预测因子,预测年龄的RMSE为7.33±1.62,MAE为5.27±1.19,r为0.88±0.08(图3a)。然后,我们在所有ATAC-seq样本上训练最终模型,并在一个完全不同的数据集上测试其性能,该数据集由Marquez等人[22]组成,包括84个经过质量控制的样本(图3b)。我们的模型提供的预测与个体的实际年龄高度相关(r=0.78)。然而,大多数个体的年龄被高估,导致RMSE(19.72)和MAE(17.29)较大。造成这种不准确的原因可能是Marquez等人使用了不同的ATAC-seq协议(原始ATAC-seq协议[27]与omoni - atac协议[33]相反)以及样本群体的遗传背景不同。我们认为,扩展训练数据集以包括以不同方式生成的样本将提高时钟对批处理效应的弹性。我们还询问了我们的时钟是否能够检测到健康状况的影响。Giroux等人先前的研究收集了SARS-CoV-2患者和健康对照者外周血的ATAC-seq数据[34]。我们在Marquez等人和本研究的样本上训练了一个额外的时钟,并比较了SARS-CoV-2阴性和阳性个体的预测年龄和实际年龄之间的差异。我们发现这种差异在阳性患者中更大(t检验,p=0.006)。我们注意到,在图3a和b中,时钟倾向于高估年轻人的年龄。这可能会混淆阳性和阴性患者的比较。因此,我们重复了SARS-CoV-2阳性和阴性个体的比较,同时使用线性模型考虑了实际年龄对时钟准确性的影响:感染的影响仍然显著,并将患者的预测年龄增加了5年(SARS-CoV-2 +效应=5.35,p=0.005,图3c)。
图3
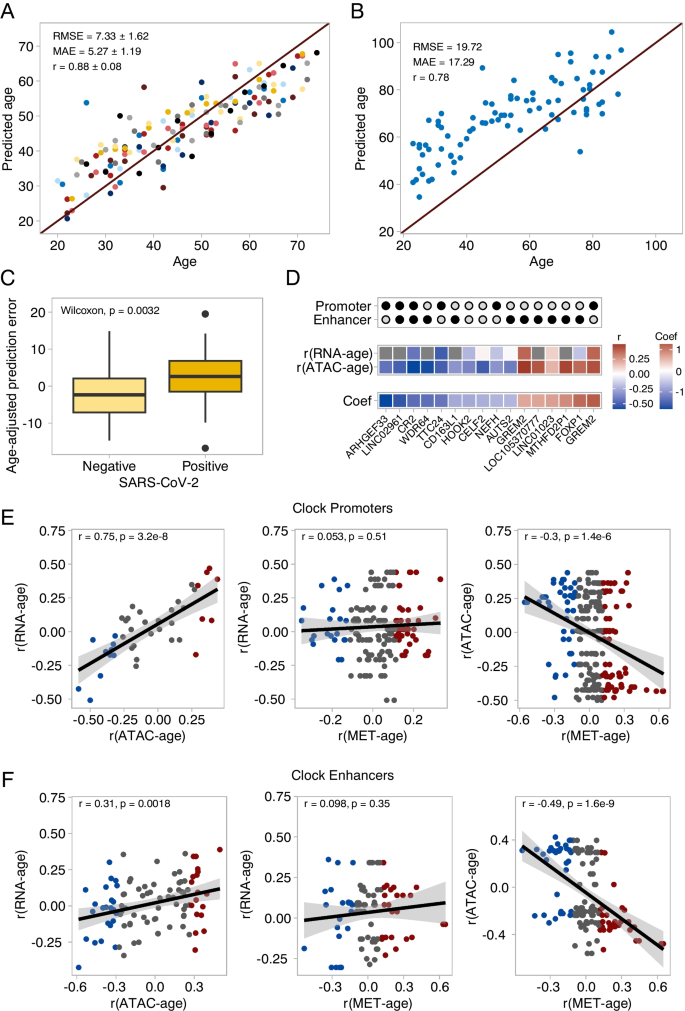
染色质可接近性预测年龄。(A)染色质可及性时钟的年龄预测。散点图显示了嵌套交叉验证的每个外部折叠的测试集预测(11个不同的模型,每个样本在测试集中一次)。均方根误差(RMSE)、绝对误差中位数(MAE)和Pearson相关系数(r)的均数和标准差均显示出来。(B)在我们所有的染色质可及性数据上训练的时钟的年龄预测,并在Marquez等人的外部数据集上进行了测试。(22)显示RMSE, MAE和Pearson’s r。(C) SARS-CoV-2阳性和阴性患者的年龄预测。对预测误差进行了调整,以解释年轻人与老年人相比对年龄的高估。未调整预测的统计检验包括在正文中。(D)最终模型中绝对系数较高的16个ocr的注释。显示了每个OCR/基因对的染色质可及性与基因表达水平的时钟系数和年龄相关性。(E)基因表达、染色质可及性和启动子DNA甲基化变化之间的关系。(F)基因表达、染色质可及性和生物钟选择的增强子DNA甲基化变化之间的关系
接下来,我们研究了最终模型的特征性质:总共选择了228个ocr,其中116个为正系数,112个为负系数。在模型选择的所有ocr中,7.5%被标注为启动子和增强子,19.3%被标注为启动子,57.0%被标注为增强子,16.2%未标注。有趣的是,时钟位点没有显示增强子的富集(Fisher精确检验,优势比=1.06,p=0.73)。这与我们在年龄相关ocr组中看到的增强子富集形成对比。一个可能的解释是,弹性网络模型不是简单地根据它们与响应变量的相关性来选择特征,而是旨在消除冗余特征。然后,我们分析了时钟选择的ocr的可及性与基因表达之间的关系。正如我们的全基因组分析所预期的那样,我们发现ocr可达性的年龄相关性与各自下游基因(启动子和增强子)的转录年龄相关性之间存在很强的相关性(图3e, f)。这表明生物钟选择的染色质可达性特征可以直接与转录组学变化及其相关的生物学过程相关。
有了这些知识,我们研究了具有最大绝对系数的时钟ocr(图3d)。这些ocr同时是GREM2的启动子和增强子,GREM2是一种编码衰老相关分泌表型(SASP)因子的基因,已知该因子与脂肪组织和皮肤的衰老有关[35,36]。在每个嵌套交叉验证模型中也选择了GREM2的启动子,突出了其预测年龄的稳健性。在我们的数据中,GREM2启动子(Spearman’s r=0.44, q=1.23e-5)和GREM2增强子(Spearman’s r=0.5, q=3.83e-7)都随着年龄的增长而打开,并与转录增加相关(Spearman’s r=0.39, q=2.1e-4)。CR2,编码补体受体2型的基因,先前已被证明在b细胞中随着年龄的增长而下降,并与缺血性中风、自身免疫性疾病和慢性感染有关[37,38]。在我们的数据中,CR2启动子/增强子的染色质可及性随着年龄的增长而显著降低(Spearman’s r=-0.59, q=2e-10),这与CR2表达(Spearman’s r=-0.42, q=3.52e-5)一致。
接下来,我们寻找我们的时钟和之前发表的基于甲基化的衰老时钟之间的相似之处。Hannum时钟[6]是在全血基础上训练的,其预测基于71个CpGs。我们的时钟包括两个跨越汉纳姆时钟位点的ocr: ARHGEF33的启动子和KLF13的增强子。有趣的是,ARHGEF33的启动子是我们时钟中系数最强的特征(-1.79)。霍瓦特泛组织时钟[7]的预测基于353个CpGs,但这些都不存在于我们的时钟选择的ocr中,这可能是因为霍瓦特时钟是针对多种组织而不仅仅是血液进行训练的。虽然我们的可及性时钟的位点很少包含Hannum和Horvath时钟使用的CpG位点,但我们研究了数据中观察到的可及性的年龄相关变化是否可以归因于DNA甲基化的变化。我们专注于汉纳姆的数据集,因为这也是从血液样本中获得的。在Illumina Infinium 450 Human Methylation array中的485,577个CpG标记中,168,778个位于我们的ocr中(123,266个位于启动子中,22278个位于增强子中,14,501个位于同时注释为启动子和增强子的ocr中,3859个位于未注释的ocr中)。我们发现,在我们的时钟选择的增强子中,衰老过程中可及性的增加与甲基化的减少相关,反之亦然(Pearson’s r=-0.49, p=1.7e-9,图3f)。这种模式也出现在我们的时钟选择的启动子中,但程度较小(Pearson’s r=-0.30, p=1.4e-6,图3e)。尽管可及性的变化与转录和甲基化之间存在关联,但甲基化的变化与启动子(Pearson’s r=0.053, p=0.51,图3e)和增强子(Pearson’s r=0.098, p=0.35,图3f)的转录变化都没有直接关系。
因此,尽管我们的时钟与Hannum时钟共享的位点很少,与Horvath时钟没有共享,但似乎与年龄相关的可及性变化可能部分与甲基化变化有关。考虑到DNA甲基化是促成染色质抑制的表观遗传机制之一,这反过来又反映在染色质可及性中,这并不奇怪。尽管如此,我们认为我们的生物钟用来预测年龄的染色质可接近性的变化不太可能完全取决于DNA甲基化。相反,可接近性的变化与基因表达的变化相关,但甲基化的变化却没有,这一事实表明,大多数与年龄相关的可接近性改变可能是两个重叠过程的结果:一个直接影响转录(可能是染色质重塑)和DNA甲基化。
我们想比较基于染色质可及性的衰老时钟和基于基因表达的衰老时钟的预测能力。因此,我们使用来自供体的样本,我们获得了ATAC-seq和RNA-seq图谱来构建两个独立的时钟(图4a)。在这个直接比较中,染色质可及性时钟在两个指标上的表现明显更好(染色质可及性时钟的RMSE=7.71±1.13,MAE=6.00±1.42,r 0.86±0.05),而基因表达时钟的RMSE=9.33±1.24,MAE=6.54±1.91,r=0.78±0.07,双尾t检验:p值=0.005 (RMSE), 0.46 (MAE), 0.005 (r),图4b)。然后,我们使用连接的染色质可及性和基因表达数据(80,400个ocr + 16,155个基因,图4a)训练了第三个“多组”时钟。该多组时钟预测年龄优于转录组时钟,但与染色质可及性时钟具有相似的准确性(RMSE=7.55±1.4,MAE=5.61±1.64,r=0.87±0.06,图4b)。此外,在所有样本上训练的最终多组时钟显示出对染色质可接近性特征的偏好,而不是转录组特征,尽管不显著(选择的ocr: 281,选择的基因:41,Fisher精确检验:优势比=1.38,p=0.06)。此外,多组时钟选择的ocr比时钟选择的基因具有更大的系数(图4c)。
图4
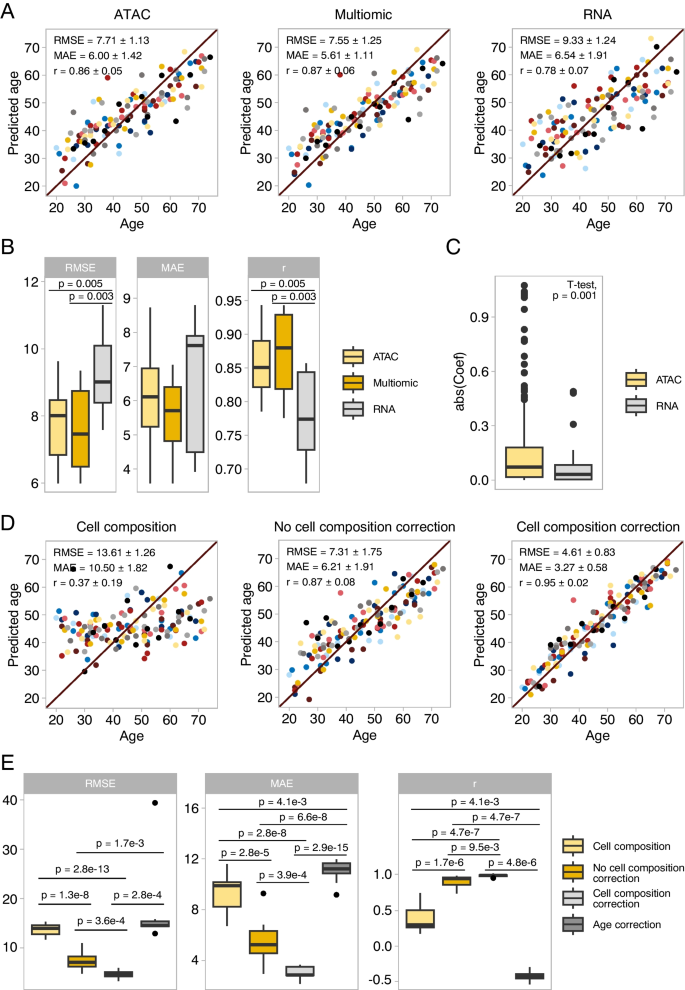
染色质可及性比基因表达更能预测年龄。校正细胞组成可提高时钟精度。(A)匹配样本上染色质可及性、转录组和多组时钟的年龄预测(n=132)。显示RMSE、MAE和Pearson’s r。(B)染色质可及性、转录组和多组时钟的评分比较。内箱形图描绘了中位数以及第一和第三个四分位数,其须状图延伸至1.5 ×四分位数范围。p值采用双尾t检验计算。(C)多组时钟选择的基因和OCR特征的绝对系数。在时钟训练之前,特征被标准化,使基因表达和染色质可接近性特征达到相同的尺度(D)单独训练细胞组成的时钟的年龄预测,不进行细胞组成校正的染色质可接近性,以及进行细胞组成校正的染色质可接近性(n=142)。(E)细胞组成、染色质可及性和校正后的染色质可及性时钟的评分比较
因此,在我们的数据集中,可访问性特征似乎比基因表达特征更能预测年龄。然而,我们注意到,通过对样本数量与我们相似的数据集中的特征进行二值化,比特年龄时钟能够从基因表达数据中获得更好的性能:n=131, RMSE=8.41, MAE=5.24, r=0.96[17]。最后,与单独使用染色质可及性相比,同时使用染色质可及性和转录组学数据来预测年龄并不能显著提高表现,但可能有助于解释生物钟。
先前的出版物报道了女性和男性在免疫衰老方面的重要差异[22]。我们观察到,与男性相比,我们的染色质可及性时钟倾向于低估女性的年龄(t检验:p=0.051,补充图6a)。因此,我们想知道纠正性别是否会改善生物钟的表现。经过性别校正数据训练的时钟并不比未经性别校正的时钟表现更好(补充图6b、c)。因此,性别差异似乎对时钟性能没有太大影响。
最后,由于我们的流式细胞术数据揭示了某些免疫细胞群的大小与年龄之间的相关性,我们想要了解ATAC时钟的性能在多大程度上取决于细胞组成的变化,而不是细胞内在的可及性变化。为此,我们使用142个样本来训练时钟,我们同时拥有染色质可及性和细胞组成数据(图4d)。仅根据细胞组成训练的时钟表现很差(RMSE=13.61±1.26,MAE=10.50±1.82,r=0.37±0.19)。此外,当我们在细胞组成和染色质可及性特征上训练时钟时,嵌套交叉验证选择的11个模型中没有一个使用细胞组成特征作为预测因子。然而,这并不排除可访问性特征可以携带有关单元组成的信息。因此,我们研究了纠正染色质可及性对细胞组成变化的影响。在细胞组成校正数据上训练的时钟(RMSE=4.61±0.83,MAE=3.27±0.58,r=0.95±0.02,图4e)比在相同未校正数据上训练的时钟序列(RMSE=7.31±1.75,MAE=6.21±1.91,r=0.87±0.08)更准确。相反,当我们校正不能用细胞组成变化来解释的染色质可及性变化时,我们再次得到了一个性能很差的时钟(RMSE=16.89±7.51,MAE=12.59±0.96,r=-0.16±0.22)。因此,细胞组成的可变性似乎对时钟性能有负面影响,即使某些细胞群大小与年龄相关。虽然校正细胞组成产生了令人印象深刻的性能,但我们注意到,这不是使用时钟的现实情况:细胞组成校正需要知道样品的年龄,以分离细胞的内在和外在影响。为了看看在没有年龄信息的情况下细胞组成校正是否可行,我们尝试在不保留年龄影响的情况下校正细胞组成,但这产生了一个非常不精确的时钟(补充图6d)。或者,我们尝试估计训练集上的校正系数,并使用它们在嵌套交叉验证中对测试集应用校正,但与未校正数据上训练的时钟相比,时钟性能没有显着提高(补充图6d)。
因此,尽管某些细胞群的大小与年龄之间存在轻微的相关性,但细胞组成本身不足以准确预测年龄。相反,细胞组成的变化似乎通过向染色质可及性数据引入噪声而降低了时钟的准确性。这种噪音可能反映了环境的影响,比如最近接触病原体,这可能部分掩盖了细胞组成中与年龄相关的变化。不幸的是,与未校正的数据相比,在没有年龄信息的情况下对细胞组成进行校正并没有提高时钟性能。尽管如此,更大的训练数据集可以更好地估计校正系数。在这种情况下,性能的提高将需要是实质性的,以证明在执行ATAC-seq之上收集流式细胞术数据是合理的。
摘要。
背景
结果
讨论
结论
方法
数据可用性
参考文献。
作者信息
道德声明
# # # # #
表观遗传时钟的一个主要限制在于其难以解释[39]。特别是,据报道,CpG甲基化与年龄相关的变化与下游基因的转录相关性较差,因此很难在甲基化改变与细胞功能破坏之间建立联系[18]。为了解决这个问题,我们研究了染色质可及性作为一种新的衰老生物标志物的适用性。
首先,我们分析了衰老对整体和局部染色质可及性的影响。我们发现与年龄相关的变化大多是局部的,并且优先影响增强剂。可及性缺乏显著的全球变化与衰老的异染色质丢失理论以及抑制组蛋白标记(如H3K9me3和H3K27me3)丢失的一些观察结果形成对比[24,25]。值得注意的是,ATAC-seq仅允许可及性的相对定量,因此我们的方法无法检测到染色质可及性的全局、均匀增加。然而,我们没有看到重复元素可达性的显著增加,TSS可达性曲线的变化,或表观遗传景观的平坦化,这可以通过trip的减少来表明。因此,术语“异染色质再分配”可能更适合描述表观基因组在衰老过程中所经历的变化。至于为什么增强子在衰老过程中更容易改变可及性,我们假设增强子的可及性可能更具动态性,因此更容易受到细胞环境局部或系统变化的影响。未来的研究可能会调查哪些染色质因子(组蛋白变异、翻译后修饰、转录因子等)驱动了观察到的可及性变化,特别是在增强子上。
接下来,我们研究了基因转录与相应调控元件染色质可及性之间的联系。总的来说,年龄相关的转录和可及性变化与相似的生物学过程有关,在位点特异性水平上,我们发现启动子和增强子的可及性变化与衰老过程中连贯的转录反应有关。相比之下,DNA甲基化对基因表达的影响较弱,这与之前的研究结果一致[18]。因此,与DNA甲基化时钟相比,染色质可及性时钟与细胞功能的联系更清晰,从而提供更好的可解释性。
然后,我们首次发现pbmc的染色质可及性谱可以用来预测供者的年龄,RMSE为7.33岁,MAE为5.27岁,r为0.88。重要的是,我们使用嵌套交叉验证来训练和验证我们的时钟,这意味着用于评估时钟性能的测试集不包括在训练和超参数调优过程中,从而导致无偏的性能估计。时钟准确地预测了年龄,尽管我们希望通过向训练集中添加更多的样本可以进一步提高性能,因为最先进的甲基化时钟通常已经在数千个样本上进行了训练。当我们用不同的ATAC-seq协议生成的先前发表的数据测试我们的时钟时,我们发现年龄预测与实足年龄高度相关,但往往高估了实际值。我们建议任何有兴趣使用我们的时钟或进行ATAC-seq的研究人员使用Omni-ATAC协议,因为Omni-ATAC提供更高质量的数据,部分原因是减少了线粒体DNA污染[33]。不幸的是,我们的工作受到了ATAC-seq数据与报告年龄信息的稀缺性的限制。我们相信,随着更多的数据可用,ATAC时钟可以训练,以更好地容忍协议和批处理效果的差异。我们在另一个公共数据集上测试了我们的时钟,该数据集包括SARS-CoV-2阳性和阴性个体的ATAC-seq数据,发现感染与更高的年龄预测相关。最近的一项研究发现,使用甲基化时钟预测年龄也有类似的短暂增加[40]。没有匹配的细胞组成数据,我们无法得出结论,这种影响主要是细胞内在的还是由感染期间循环细胞群的变化驱动的。尽管如此,这可能表明SARS-CoV-2感染引起的全身性炎症与炎症相似:炎症标志物水平的慢性年龄相关增加,包括细胞内在和组成性免疫失调[29,41]。
由于我们已经匹配了ATAC-seq和RNA-seq谱,我们可以直接将染色质可及性时钟的性能与转录组对应物进行比较。在这种直接比较中,我们的atac时钟表现明显更好。当我们进一步开发基于染色质可及性和转录组特征的多组时钟时,我们发现多组时钟的表现与可及性时钟相似,并且更多地依赖于可及性特征而不是基因表达特征,这再次表明染色质可及性数据可能比转录组数据更能预测年龄。基因表达的变化可能比染色质可及性更快(例如,对压力的反应,昼夜节律调节等),因此在预测中引入了更多的噪声。
最后,我们研究了细胞组成变化的相对重要性,而不是细胞内在的可及性变化,发现细胞组成校正提高了时钟的性能。细胞组成可能取决于最近与病原体的接触,这解释了为什么校正细胞组成可能是有益的。
我们已经证明了基于染色质可及性的表观遗传时钟的可行性,它与转录有很强的关系,同时比转录组时钟表现得更好。我们希望这将为该领域提供一种制造可解释老化时钟的新方法。
来自159名年龄在20至74岁之间的献血者的匿名全血从瑞士洛桑-埃帕林的跨区域输血中心获得。内部审查委员会批准了这项研究,所有献血者都书面同意将他们的血液用于研究目的。血样采集后4.5 h内处理。
血液用等量的Dulbecco磷酸盐缓冲盐水(Gibco)稀释,并分层在Histopaque-1077 (Sigma-Aldrich)上。根据制造商的方案进行密度梯度离心,收集并洗涤pbmc。在LUNA-II自动细胞计数器(Logos Biosystems)上计数细胞,并立即进行ATAC-Seq文库制备、RNA提取和PBMC染色/固定。所有协议同时进行。
根据Omni-ATAC协议[33],使用EPFL Protein Production and Structure facility提供的Tn5进行ATAC-Seq文库制备。转置片段使用MinElute PCR纯化试剂盒(Qiagen)纯化。利用2 × NEBNext Master Mix (NEB)和具有Illumina测序(IDT)独特双指标的预混合引物对洗脱液进行PCR扩增。文库采用SPRIselect (Beckman Coulter)双面珠粒选择纯化。
按照制造商的方案,使用Monarch Total RNA Miniprep Kit (NEB)进行RNA提取。
细胞采用Ghost-Dye/V510、CD3 + /V421、CD4 + /FITC、CD8 + /APC- cy7、CD16 + /PE、CD19 + /PE- cy7和CD56 + /APC (Biolegend)染色。细胞在固定和渗透溶液(fixed and Permeabilization Solution, BD)中固定。采用Cytoflex S流式细胞仪(Beckman Coulter)分析亚群比例。
ATAC-Seq文库在Novogene (UK) Company Limited的Illumina NovaSeq 6000上进行150 bp的配对端测序,测序深度为3000万reads。原始读数是适配器和质量修剪使用修剪Galore![42]并使用蝴蝶结2(设置-非常敏感-X 1000 -燕尾)映射到人类基因组的GRCh38构建[43]。在调用峰值之前,使用samtools[44]和Picard工具[45]对原始样本进行过滤,去除具有多个映射、PCR重复和线粒体读取的reads。
BAM格式的对齐被转换为BED,并使用MACS2调用峰值(设置-f BED -g“hs”-keep-dup“all”-q 0.01 - nommodel -shift -100 -extsize 200)[46]。为了定义一个共同的峰集,我们首先在窄峰MACS2输出上使用BEDTools merge计算所有单独峰集的并集[47]。接下来,我们使用BEDTools multiinter识别出在多个样本中被可靠地称为峰值的区域,并将输出过滤到在50个或更多样本中被称为峰值的区域。然后,我们使用BEDTools intersect过滤联合峰集,使其只包含至少一个可靠调用区域的峰。最后,我们丢弃了与ENCODE黑名单重叠的峰[48]。
我们使用featurecots[49]生成了一个原始计数表,具体计数Tn5切割位点,而不是整个片段。首先通过将每个峰值的计数除以峰值的长度(以千为单位)将计数转换为读取密度,然后通过除以峰值中读取的总数(以百万为单位)将计数归一化。
我们认为质量好的比对少于1100万次和/或trip低于0.18的样品为低质量样品。此外,我们在对数尺度的归一化计数的前两个主成分上使用椭圆包络法丢弃了样本中的异常值。这除去了16个样本,包括所有低测序深度和低trip的样本。
用于测试ATAC时钟的Marquez等人[22]和Giroux等人[34]数据库中的样本从原始读取开始处理,随后作为我们自己的样本进行映射和过滤。但是,没有执行峰值调用,而是在我们的数据单独生成的峰值集上计算读取。与我们自己的数据集一样,我们删除了异常值,但我们没有施加最小的trip,因为我们没有对这些样本执行峰值调用。
覆盖bigWig轨道是使用deepTools bamCoverage[50]从以Tn5切割位点为中心的区域生成的,其缩放因子与用于规范化计数的缩放因子相同。使用deeptools computemmatrix和plotProfile从大型轨道生成TSS剖面。
RNA-Seq文库制备和测序由Novogene (UK) Company Limited在Illumina NovaSeq 6000上以150 bp对端模式进行。使用FastQC评估原始FASTQ文件的质量、适配器内容和重复率。使用STAR比对器(v2.7.9a)[51],用' -sjdbOverhang 100'将Reads与人类基因组(GRCh38)进行比对。每个基因的reads数使用subread包中的featurements函数进行量化[49]。使用AnnotationDbi包[52]和org. hs . egg .db包[53]中的mapIds函数将Ensembl转录本映射到基因符号上。EdgeR使用m值法的修剪方法对行数进行归一化,并过滤低表达基因[54]。最后,使用与ATAC-seq数据集相同的策略去除15个异常值。我们使用相同的管道处理Marquez等人的RNA-seq数据。
弹性网络模型的训练和验证在Python中使用Scikit-learn模块进行[55]。特征在训练前使用StandardScaler进行标准化。将样本分为11组,使每组的年龄构成均匀覆盖年龄范围。使用嵌套交叉验证来调整超参数并估计模型的性能。外部和内部交叉验证循环都作为“留一组”交叉验证运行,这意味着外部循环使用11组中的每一组一次作为测试集,而内部循环在剩余的10组上交替进行。使用均方根误差(RMSE)、中位数绝对误差(MAE)和Pearson相关系数(r)来报告模型的性能。每当多个时钟相互比较时,它们都会在来自相同供体的样本上进行训练,并使用相同的分区进行交叉验证。在使用oom[57]准备数据后,使用limma软件包中的removeBatchEffect在R中对性别和细胞组成进行校正[56]。除非另有说明,年龄信息被纳入实验设计以保持年龄效应。当校正细胞组成作为时钟训练的一部分时,我们使用MultiOutputRegressor与线性回归拟合模型,将训练集中每个ocr的可访问性解释为年龄和细胞组成的函数。然后使用细胞组成特征的拟合系数从训练集和测试集中减去细胞组成的影响。
如果ocr位于转录起始位点的上游或下游1000 bp,则将其标记为启动子。如果ocr与PEREGRINE数据集中标注为增强子的区域重叠,则ocr被标注为增强子[31]。值得注意的是,我们允许将ocr标注为启动子和增强子。在启动子的情况下,ocr与最近的基因连接,而对于增强子,我们测试了与最近的基因连接或使用PEREGRINE数据集中的基因-增强子链接。基因组的重复区域使用重复掩模进行鉴定[58]。
在R中进行统计分析。GSEA使用ClusterProfiler R包进行1000个排列[59]。在对ATAC-seq数据执行GSEA时,我们按照以下步骤创建了一个基因排序列表:1)从如上所述产生的基因- ocr链接开始,我们丢弃没有链接基因的ocr; 2)我们丢弃不表达基因的ocr基因对。3)当一个基因连接到多个ocr时,我们选择了染色质可及性和表达在我们的ATAC-seq和RNA-seq数据共有的一组样本中最相关的ocr基因对。这就产生了1:1的ocr基因链接。另外,我们只选择启动子基因对,这也产生了1:1的链接。4)我们根据染色质可及性与年龄之间的spearman相关性对基因进行了排序。
ccDownload: /内容/ pdf / 10.1007 / s11357 - 023 - 00986 - 0. - pdf

